Redox Biology and Diseases
Topics

Thiol-redox metabolism in the endoplasmic reticulum (ER): the redox control of secretion
Leaders: Michel B. Toledano and Agnès Delaunay-Moisan
The endoplasmic reticulum (ER) is the gateway to the secretory pathway. Secreted and membrane proteins are translocated into the ER, glycosylated, and folded before continuing their route to their destination. Protein misfolding causes ER stress, which activates the Unfolded Protein Response (UPR) as an adaptive measure. When adaptation fails, chronic ER stress leads to metabolic dysfunctions, abnormal H2O2 production, and cell death, which contributes to the pathophysiology of major diseases such as diabetes.
The formation of disulfide bonds in the ER is crucial for the folding and stabilization of many secreted proteins (Fig. 1, [a-c]). The conserved ER oxidase Ero1, a flavoenzyme, is responsible for the de novo generation of protein disulfide bonds (Fig. 1, [a, b]). Ero1 uses O2 as electron acceptor to form a disulfide bond, thus producing H2O2 as by-product. The disulfide bond generated by Ero1 is then transferred to protein disulfide isomerase (PDI). Oxidized PDI then transfer the disulfide bond to a substrate (Fig. 1, [a]).
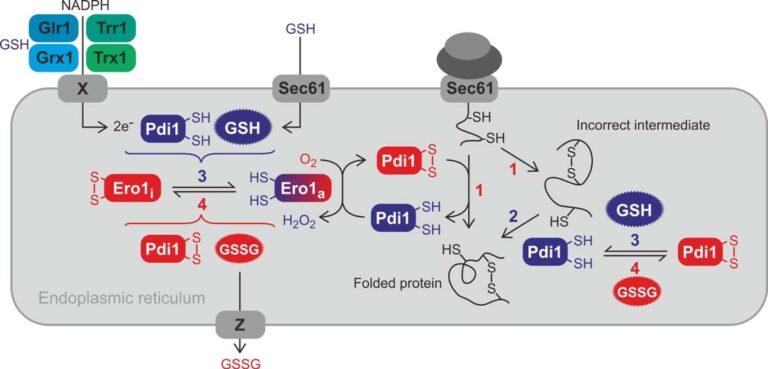
Figure 1. Overview of the ER redox network and its relevance for oxidative protein folding. Proteins are oxidized in the ER by oxidized PDI with the help of active Ero1a (1). Incorrect disulfide bonds in misfolded proteins are isomerized (i.e. reduced and reoxidized) by PDI (2). Regulatory disulfide bonds are formed in inactivate Ero1i. The formation and reduction of the regulatory disulfide bonds of Ero1 depends on the redox state of PDI and glutathione (3 and 4). The exact mechanisms remain to be elucidated. GSH is imported by Sec61. Electrons for the ER reducing pathway might be provided by cytosolic NADPH with the help of oxidoreductases and an unknown transmembrane protein with DsbA-like activity (X). Whether ER GSSG is lost by vesicular transport or exported out of the ER by specific export (Z) is unknown.
An outstanding question. To achieve correct cysteines pairing, disulfide bond isomerization or cycles of reduction and reoxidation are necessary. How then a balance between thiols and disulfides bonds is maintained in the ER to allow efficient oxidation of proteins while preventing irreversible cysteine mispairing? Is there a need for an ER thiol reducing pathway to maintain this balance?
A solution to this conundrum is provided by bacteria. The periplasm of prokaryotes carries an oxidative pathway, made of DsbA, the terminal thiol oxidase, and of DsbB, a transmembrane (TM) protein functionally equivalent to Ero1, which generates disulfide bonds by giving electrons to O2 through the respiratory chain. The periplasm also carry a reducing pathway, made of DsbC, the terminal thiol reductase, and of DsbD, a TM protein reduced by thioredoxin and NADPH in the cytosol. Kinetic barriers separate, DsbA and DsbC, to allow them to operate opposite reactions simultaneously in the same compartment. In eukaryotes, however, PDI assumes both the oxidation of cysteines and the reduction of disulfides, which raises the question of the source of PDI ER reducing power.
In eukaryotes, the first model, provided by Sevier and Kaiser, does not invoke the need for net ER reducing power. These authors observed that yeast Ero1 is inhibited by regulatory disulfide formation, later also identified in mammalian Ero1 (Fig. 1[a, b]), and proposed that Pdi1-mediated Ero1 reductive activation and, reciprocally, Ero1 oxidation of Pdi1 establish a feedback control of Ero1 activity influenced the thiol-redox state of the ER through Pdi1, which acts as ER thiol-redox sensor.
Net ER reducing power is however needed in the ER, not only for oxidative protein folding, but also for the reduction of terminally misfolded substrates before their degradation by the ERAD-proteasome pathway, and for proteins that undergo reversible thiol oxidation as part of their regulation, with Ero1 and Kar2 (see below) as best relevant examples. The reduction of disulfide bonds requires NADPH as the electron donor, but the ER only contains thiol-disulfide oxidoreductases, such as PDI, and no NADPH:disulfide oxidoreductases (Fig. 1, [a]). Thus, the ER reducing pathway must be linked to either or both of the two cytosolic disulfide reducing pathways: thioredoxin consisting of the NADPH-dependent thioredoxin reductase and thioredoxin, and glutathione, consisting of NADPH-dependent glutathione reductase, glutathione, glutaredoxins (Fig. 1). Which of these two pathways is the source of the ER reducing power is not well understood, although both are presumably needed, each with a distinct function.
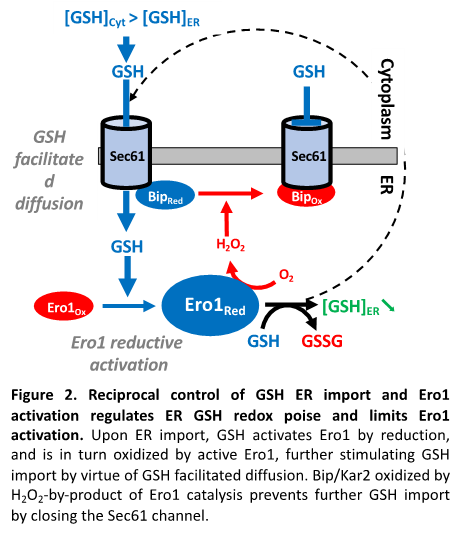
Glutathione. Glutathione is present in the ER, but whether it provides net ER thiol-reducing power, and even whether it is needed is controversial (Fig. 1 [a-c]). In both yeast and mammalian cells, ER GSH depletion does not trigger ER stress, nor does it alter basal secretion, which has casted doubts on the need for GSH in the ER. To learn on the role of GSH in the ER, our group searched for the mechanism of its import into the ER, and found the existence of a mutual control between the ER import of GSH and the activation of Ero1 (Fig. 2). We showed that the ER import of GSH proceeds through the Sec61 protein translocon (Fig. 1, [b], Fig. 2) by passive diffusion entrained by the gradient established by GSH cytosolic synthesis and GSH oxidation by the ER oxidase Ero1. Upon ER import, GSH triggers Ero1 reductive activation by reducing PDI, and reciprocally when active, Ero1 inhibits the ER import of GSH by promoting the oxidation of the chaperone Bip/Kar2 by H2O2 byproduct formation. Kar2 oxidized on its unique cysteine residue to a sulfenate (-SOH) loses ATPase activity, becoming an holdase competent to seal Sec61 permeability. Of note, upon ER import, GSH breaks Sec61 permeability, possibly allowing ER Ca2+ leak. The mutual control of the ER oxidation pathway and of net GSH ER reducing power availability provides an exciting model to how the thiol-disulfide balance is kept in the ER, which depart from the model that solely attributes this control to the regulation of Ero1
Thioredoxin.
The thioredoxin pathway is absent in the ER lumen, but paradoxically, inactivation of cytosolic thioredoxin reductase causes a constitutive UPR in yeast and prevents the maturation of disulfide-containing ER proteins in mammals, which suggests that this pathway contributes to ER oxidative protein folding. As thioredoxin is not expressed in the ER, an ER transmembrane protein, akin to periplasmic DsbD, must transfer electrons from cytosolic thioredoxin to the ER reducing systems (Fig. 1).
OUR CURRENT EFFORTS ARE:
(i) Establish the mechanism of the import of GSH in the mammalian ER and its regulation. This import appears more complex than in yeast, possibly involving other transporters in addition to Sec61.
(ii) Identify the eukaryotic functional homologue of prokaryotic DsbD
(iii) Sort out the respective functions of thioredoxin and GSH in the unstressed and stressed ER
OUR EXPERIMENTAL APPROACHES COMBINE, in addition to usual approaches of yeast and mammalian cells genetics and cell and molecular biology, including cell imaging:
(i) The extensive use of redox biochemical approaches to faithfully establish the redox state of cysteine residues in cellulo. These approaches allow to identify the nature of the oxidized form of the cysteine residue, whether a disulfide bond (P-S-S-P’), a sulfenic acid (P-SOH), a sulfinic acid (P-SO2H), or a persulfide (P-S-SH). Redox biochemistry is applied at the single protein level, and also at the proteome-wide level (redox proteomics)
(ii) The use of genetically-encoded fluorescent probes:
– To establish the redox state of GSH and measure its concentration in a compartment-specific manner
– To measure le concentration of H2O2 in a compartment-specific manner
(iii) We do not restrict ourselves to these approaches, but will initiate any new technique that appear critical to pursue our research objectives, usually through collaborations: we are now performing high throughput yeast genetic screens, mammalian chemical screens, and measuring cell Ca2+ fluxes with dedicated probes.
The mechanism of Iron-Sulfur Cluster assembly
Leader: Benoit D’Autréaux
A. RECONSTITUTION AND MECHANISM OF THE ISC MACHINERY
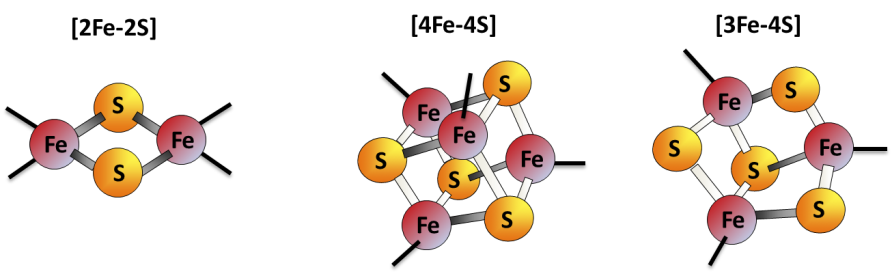
Iron-sulfur (Fe-S) clusters are protein cofactors made of iron and sulfur conserved in all life kingdoms. Fe-S cluster redox properties endow them the ability to transfer electrons, thus constituting the catalytic center of many redox enzymes and signaling proteins, and contributing to a multitude of essential cellular processes.
These processes include energy metabolism, the synthesis of proteins, the maintenance of genome integrity, and the sensing of iron. Due to their essential biological roles, any defect in the biosynthesis of Fe-S cluster leads to alterations of iron metabolism, and to severe human diseases. The most common of these diseases is the heritable Friedreich’s ataxia (FA), which causes major neurodegenerative and cardiac disorders, and is linked to impaired expression of frataxin (FXN), a protein that has a central role in Fe-S cluster biosynthesis. The high medical importance of Fe-S clusters has spurred intensive research to understand how these cofactors are biosynthesized and assembled into proteins, which is our focus of research.
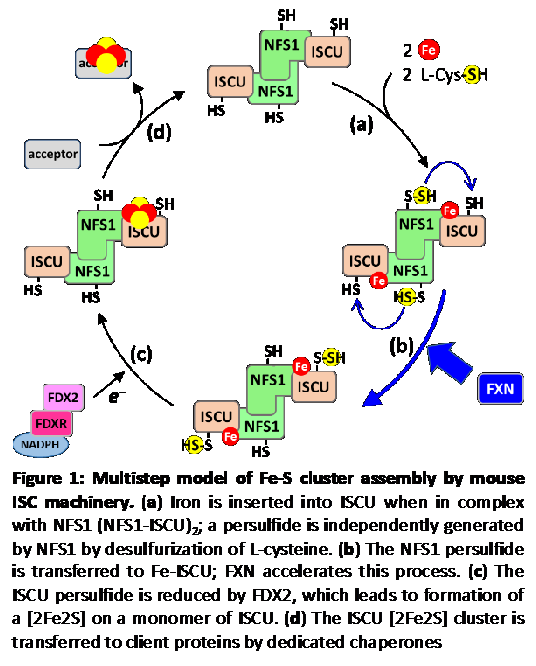
Fe-S clusters biosynthesis involves complex biochemical mechanisms operated by highly conserved multi-protein machineries. Starting a few years ago, we have begun to address these mechanisms. Our approach consisted at reconstituting the ISC assembly machinery in vitro with all its components, which comprise the scaffold protein ISCU, the cysteine desulfurase NFS1, the ferredoxin reductase complex FDX2-FDXR, and frataxin (FXN). This reconstitution allowed us the access to the all multistep process and to elucidate the function of FXN and FDX2, which were poorly understood, which provided a first sequential model of this multistep process (Fig. 1).
The model: Fe-S cluster assembly is initiated by the insertion of a ferrous iron in the assembly site of ISCU (Fig.1, step a). Surprisingly, ISCU binds iron without the need of any other proteins, suggesting that there are no iron chaperones involved, with iron probably directly originating from the pool of labile ferrous iron. NFS1, in complex with ISD11 and ACP, provides sulfur by desulfurization of L-cysteine, thereby generating a cysteine-bound persulfide on NFS1 (Cys-SSH) (Fig.1, step a). The persulfide is then transferred to ISCU, only when the latter is already bound by iron (Fig.1, step b). The ferredoxin reductase complex, FDX2-FDXR, then reduces the persulfide into sulfide, which leads to formation of a [2Fe2S] cluster (Fig.1, step c), eventually transferred to client proteins (Fig.1, step d). FXN accelerates the whole biosynthetic process by increasing the rate of persulfide transfer from NFS1 to ISCU (Fig.1, step b).
By establishing that FXN accelerates persulfide transfer from NFS1 to ISCU (Fig. 1, step 1), we ruled out previous claims that this enzyme is required for iron insertion on ISCU or for persulfide formation. By finding that both persulfide transfer and its reduction only occurs when ISCU is already bound by iron (Fig. 1, steps 2 and 3), indicates that sulfur insertion on ISCU is coordinated with the loading of iron on ISCU. We also undertook a structural characterization of the iron center in the ISCU assembly site by combining several biophysical methods (EPR, NMR, XAS and Mössbauer spectroscopies), and found that the ISCU Fe++ center is mononuclear, coordinated by four strictly conserved amino acids in the assembly site: Cys35, Asp37, Cys61 and His103 with the receptor for the persulfide emanating from NFS1, Cys104, at close proximity. We claim that our model is the first to describe a truly enzymatically catalyzed, and therefore physiologically relevant process, unlike previous models. In particular, previous reconstitutions used thiols (DTT, GSH, L-cysteine) instead of FDX2 as the reductant, which reduce the persulfide on NFS1, and not on ISCU as FDX2 does, which causes release of “free” sulfide, irrespective of the presence of iron, and explains that under these conditions, ISC assembly is poorly efficient, probably akin to a spontaneous chemical reconstitution, and therefore not physiologically relevant.
OUR CURRENT EFFORTS FOCUS ON:
(i) Elucidate the mechanisms of steps b and c that ensure the coordination of iron and sulfur assembly; how escape of the sulfide ions is prevented upon reduction by FDX2; how a binuclear [2Fe2S] cluster is formed from a mononuclear iron center in ISCU?
(ii) Elucidate the mechanism of how FXN stimulate persulfide transfer, as a major asset for the design of FXN-substituting drugs for the treatment of FA
(iii) Elucidate the molecular details of the sequential actions of FDX2 and FXN
OUR EXPERIMENTAL APPROACHES
As of today, our work uses in vitro enzymatic approaches after reconstitution of the ISC machinery with purified proteins that are mostly performed in an anaerobic chamber, due to the extreme redox lability of iron in ambient air, identify reaction intermediates of the ISC assembly process at the molecular and atomic levels using an array of complementary and advanced biochemical, structural and spectroscopic methods to trap and characterize reaction intermediates.
B. SEARCH FOR THERAPEUTIC MOLECULES THAT SUBSTITUTE FOR FRATAXIN
Based on our reconstitution of the ISC assembly machinery, and elucidation of the role of FXN in stimulating persulfide transfer from NFS1 to ISCU, we established a very simple in vitro assay of FXN function that measures the efficiency of the all multistep process to screen for chemicals that bypass the need for FXN in ISC assembly.