Biochemistry of Metalloproteins
and Associated Diseases
Topics
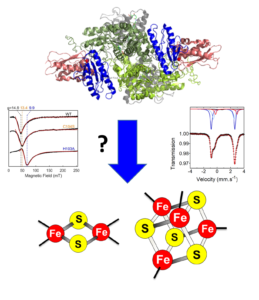
Mechanism of Fe-S cluster biogenesis
Fe-S clusters biosynthesis involves complex biochemical mechanisms operated by highly conserved multi-protein machineries. This process is initiated in mitochondria in the which the Iron-Sulfur Cluster (ISC) assembly machinery synthesizes [2Fe2S] clusters that are the building blocks for the synthesis of [4Fe4S] clusters (Fig. 1). [2Fe2S] and [4Fe4S] clusters are assembled on the ISCU and ISCA scaffold proteins, respectively, then transferred to recipient proteins by specialized accessory proteins. The core ISC machinery also synthesizes a compound that is exported to the cytoplasm by the ABCB7 transporter for the synthesis of Fe-S clusters of cytosolic and nuclear Fe-S proteins (Fig. 1). This compound is possibly a [2Fe2S] cluster that is the substrate of the Cytosolic Iron-sulfur cluster Assembly (CIA) machinery that assembles [4Fe4S] clusters (Fig. 1).
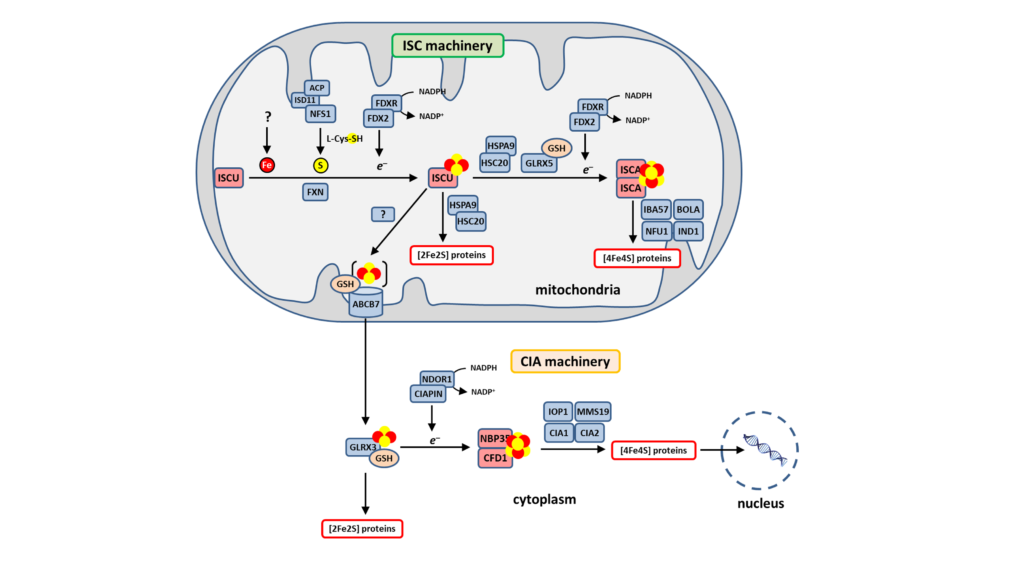
We started a few years ago to address the mechanisms of these processes. Our approach consisted at yeast genetics and in vitro reconstitution of the mitochondrial ISC machinery with all its components, which comprise the scaffold protein ISCU, the cysteine desulfurase NFS1, the ferredoxin reductase complex FDX2-FDXR, and frataxin (FXN). This reconstitution allowed us the unravel the first steps of this process and to elucidate the function of FXN and FDX2, which were poorly understood (Fig. 2). This provided a first sequential model of this multistep process.
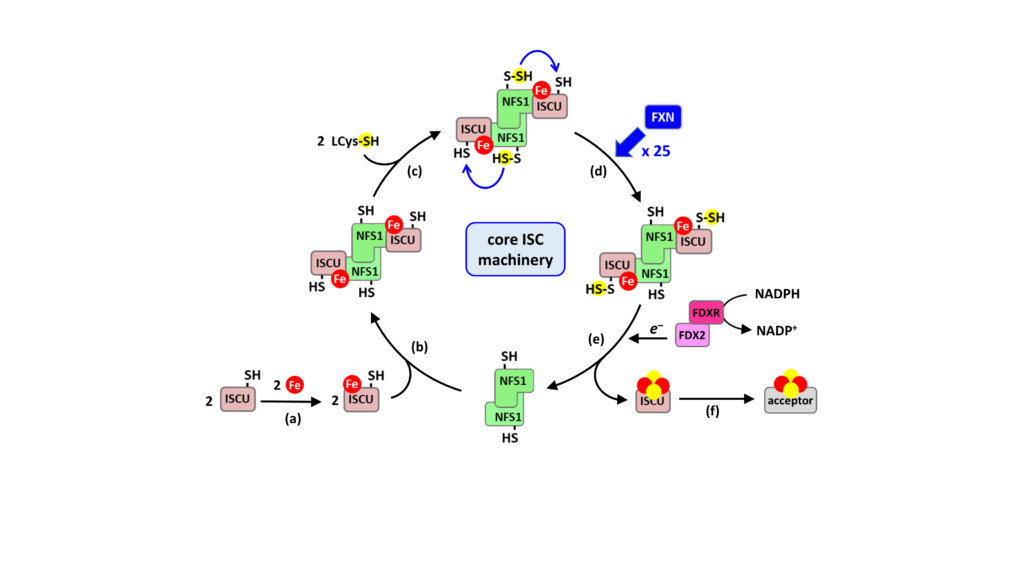
model of [2Fe2S] cluster biosynthesis by the core mitochondrial ISC machinery
Fe-S cluster assembly is initiated by the insertion of a ferrous iron in the assembly site of ISCU (step a). ISCU binds iron without the need of any other proteins, suggesting that there are no iron chaperones involved, with iron probably directly originating from the pool of labile ferrous iron. ISCU binds to NFS1 (step b), which in complex with ISD11 and ACP (omitted for clarity), provides sulfur by desulfurization of L-cysteine, thereby generating a cysteine-bound persulfide on NFS1 (Cys-SSH) (step c). The persulfide is then transferred to ISCU, only when the latter is already loaded with iron (step d). The ferredoxin reductase complex, FDX2-FDXR reduces the persulfide into sulfide, which leads to formation of a [2Fe2S] cluster (step e), that is subsequently transferred to client proteins (step f). FXN accelerates the whole biosynthetic process by increasing the rate of persulfide transfer from NFS1 to ISCU (step d).
We claim that this model is the first to describe a physiologically relevant process, unlike previous models. In particular, previous reconstitutions used thiols (DTT, GSH, L-cysteine) instead of FDX2 as the reductant, which reduce the persulfide on NFS1, and not on ISCU as FDX2 does, which causes release of “free” sulfide, irrespective of the presence of iron, and explains that under these conditions, ISC assembly is poorly efficient, probably akin to a spontaneous chemical reconstitution, and therefore not physiologically relevant.
We recently solved the structure of the iron center in the ISCU assembly site by combining several biophysical methods (EPR, NMR, XAS and Mössbauer spectroscopies), and found that the iron center is mononuclear, holding a Fe2+ ions coordinated by four strictly conserved amino acids of the assembly site: Cys35, Asp37, Cys61 and His103 with the receptor for the persulfide emanating from NFS1, Cys104, at close proximity. These data provides the first step toward the elucidation of the whole Fe-S cluster assembly sequence.
OUR CURRENT EFFORTS FOCUS ON:
(i) The source of iron for ISCU
(ii) The mechanism of step d that ensure the coordination of iron and persulfide insertion into ISCU
(iii) The mechanism of persulfide transfer acceleration by FXN, as a major asset for the design of FXN-substituting drugs for the treatment of FA
(iv) The mechanism of step e that is likely a multistep process by addressing the following questions: how escape of the sulfide ions is prevented upon reduction by FDX2; how a binuclear [2Fe2S] cluster is formed from a mononuclear iron center in ISCU?
(v) The molecular details of the sequential actions of FDX2 and FXN
(vi) The distribution of Fe-S clusters between client proteins, ISCA and ABCB7 which requires to determine the nature of the compound that is provided to and exported by ABCB7
Drug-discovery for Friedreich’s Ataxia
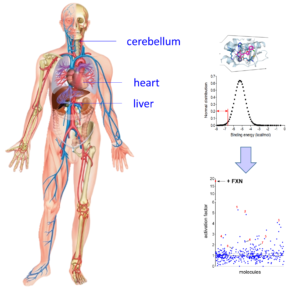
Fe-S cluster enzymes play essential roles in cellular metabolism, therefore any defect in their biosynthesis or in a specific Fe-S enzyme lead to pathological conditions such as anemia, neurodegeneration, cardiomyopathy and cancers. The most common of these diseases is the heritable Friedreich’s ataxia (FA), a neurodegenerative and cardiac disease caused by defective expression of the frataxin protein (FXN), a central component of whole Fe-S cluster biogenesis network. FA is caused by GAA triplet expansion in the first intron of the FXN gene, which leads to silencing of transcription and consequently a dramatic decrease in the FXN protein level, up to 95 % in FA patients. FA mostly affects the tissue expressing FXN such as the heart, cerebellum, spinal cord, liver, skeletal muscle, and the pancreas, which triggers hypertrophic cardiomyopathy, cerebellar, limb and sensory ataxia and diabetes.
The biochemical function of FXN has been a persistent question since its discovery in the mid-90s. By establishing that FXN is a stimulator of Fe-S cluster that accelerates persulfide transfer from NFS1 to ISCU, the BMAD team ruled out previous claims that this enzyme is required for iron insertion on ISCU or for persulfide formation (voir Thème N°1). This has open new perspectives for the development of strategies to cure FA.
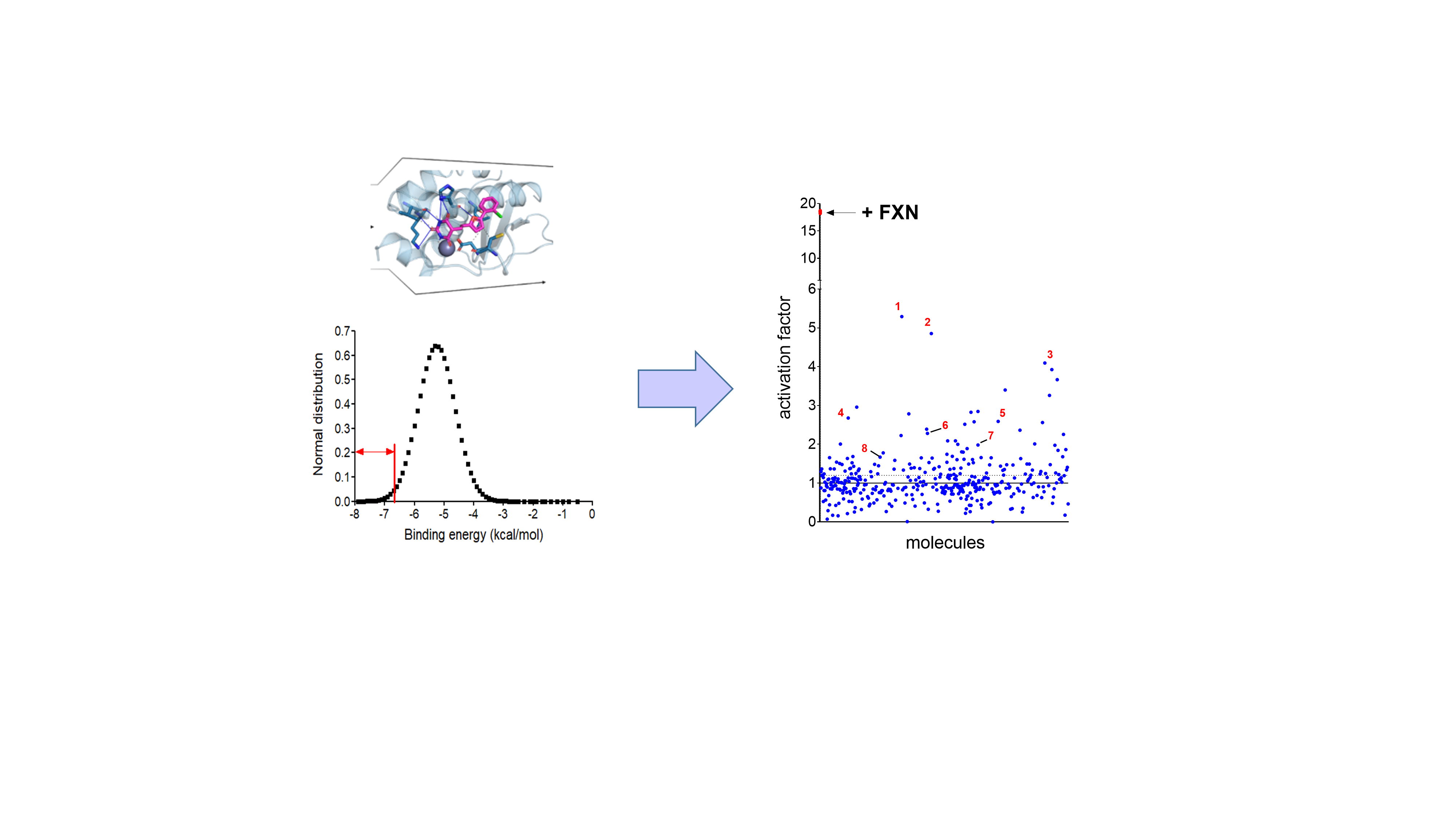
Figure 3: Virtual screening and high throughput assays for FA
Left panel: Virtual screening of the French national chemical library hold by ChemBioFrance (82,500 molecules) by docking on the FXN binding site in the structure of the NFS1-ISCu complex. Right panel: high throughput screening of the 420 molecules with the highest affinities showing activation factors.
OUR CURRENT EFFORTS FOCUS ON:
i) Elucidation of the mechanism of persulfide transfer acceleration by FXN
ii) High throughput screenings of chemical libraries to find molecules replacing FXN in persulfide transfer acceleration
iii) Virtual screening to pre-select drug candidates from chemical libraries
iv) Testing drug candidates in FA drosophila models
Functional roles of Fe-S clusters in enzymes and proteins
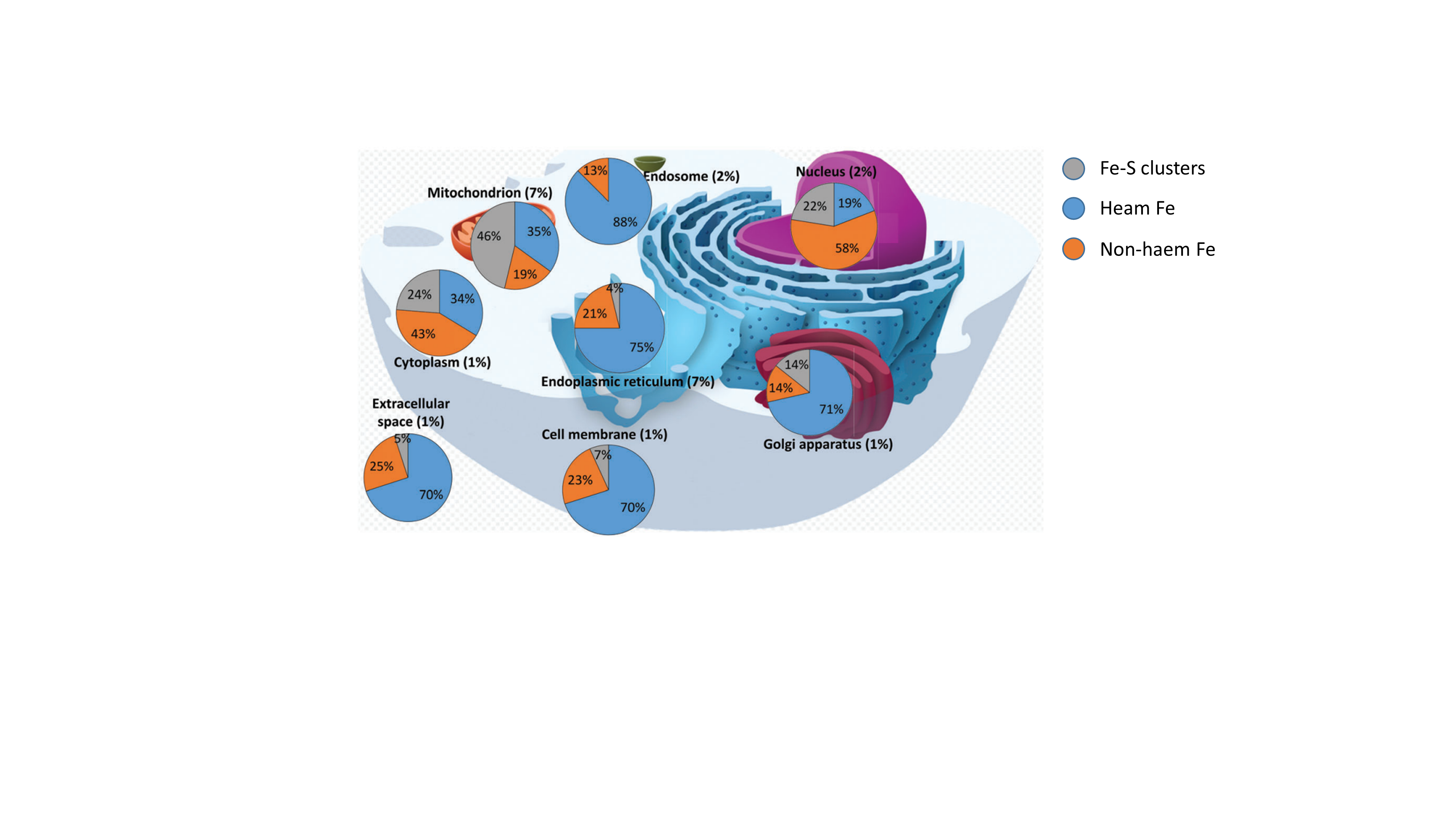
Owing to their versatile redox properties and their abilities to bind and react with small molecules and substrates, Fe-S cluster enzymes and proteins fulfill a wide range of biological functions in all type of living organisms (eukaryotes, bacteria and archaea): electron transfer for the production of ATP, redox and non-redox catalysis for the synthesis of a multitude of cellular components and metabolites, signaling and maintenance of genome integrity. They are present in most cellular compartments: mitochondria, cytoplasm, nucleus, cell membrane, endoplasmic reticulum, Golgi (Fig. 4). While canonical primary sequences exist that have allowed predictions of Fe-S cluster binding sites in proteins, which has strongly participated to the establishment of the first lists of Fe-S cluster proteins, many non-canonical sequence also exists. Thereby, the number of Fe-S cluster enzymes is constantly growing as new Fe-S cluster enzymes are discovered. In many cases, the functional role of these Fe-S clusters is not known. The BMAD team is interested in the elucidation of the functional role of newly discovered Fe-S cluster enzymes.
The team of Benoit D’Autréaux has participated to the identification of one of the very first Fe-S cluster in a viral protein in collaboration with the team of Didier Poncet (I2BC). The protein was originating from a Rotavirus strain that are a major cause of acute gastroenteritis worldwide. Rotavirus express non-structural proteins during their replication cycle that assist the packaging of the structural proteins and the double stranded RNA. The binding of a [2Fe2S] cluster by the non-structural protein NSP5 was shown to mitigate its affinity for RNA, which could possibly affect its replication cycle. Biochemical studies have shown that only two cysteines (Cys171 and Cys174) from each monomer were required for the binding of the [2Fe2S] cluster, suggesting a bridging mode between two monomer of NSP5 (Fig. 5a). Recent alphafold predictions have confirmed this topology with each cysteine tandem (Cys171 and Cys174) spatially facing the other one on the other subunit (Fig. 5b). These data have further indicated that virus are able to hijack the Fe-S cluster assembly machineries of their host to acquire a cluster, but the underlying mechanism remains unclear Today, a few other viral proteins coordinating a Fe-S cluster have been identified: the GpL protein from phage λ (Tam et al. JMB 2013), the MCPyV small T antigen protein from Polyomavirus (You et al. J. Virol. 2015) and more recently the RNA-dependent RNA-Pol from SARS-Cov2 (Maio et al. Science 2022) and the Glycine/Cysteine-Rich protein from Megavirinae Giant Virus (Villalta et al. JACS 2022). In most cases, the functional role of these clusters is not known. The BMAD team is interested in the study of the role of these clusters.
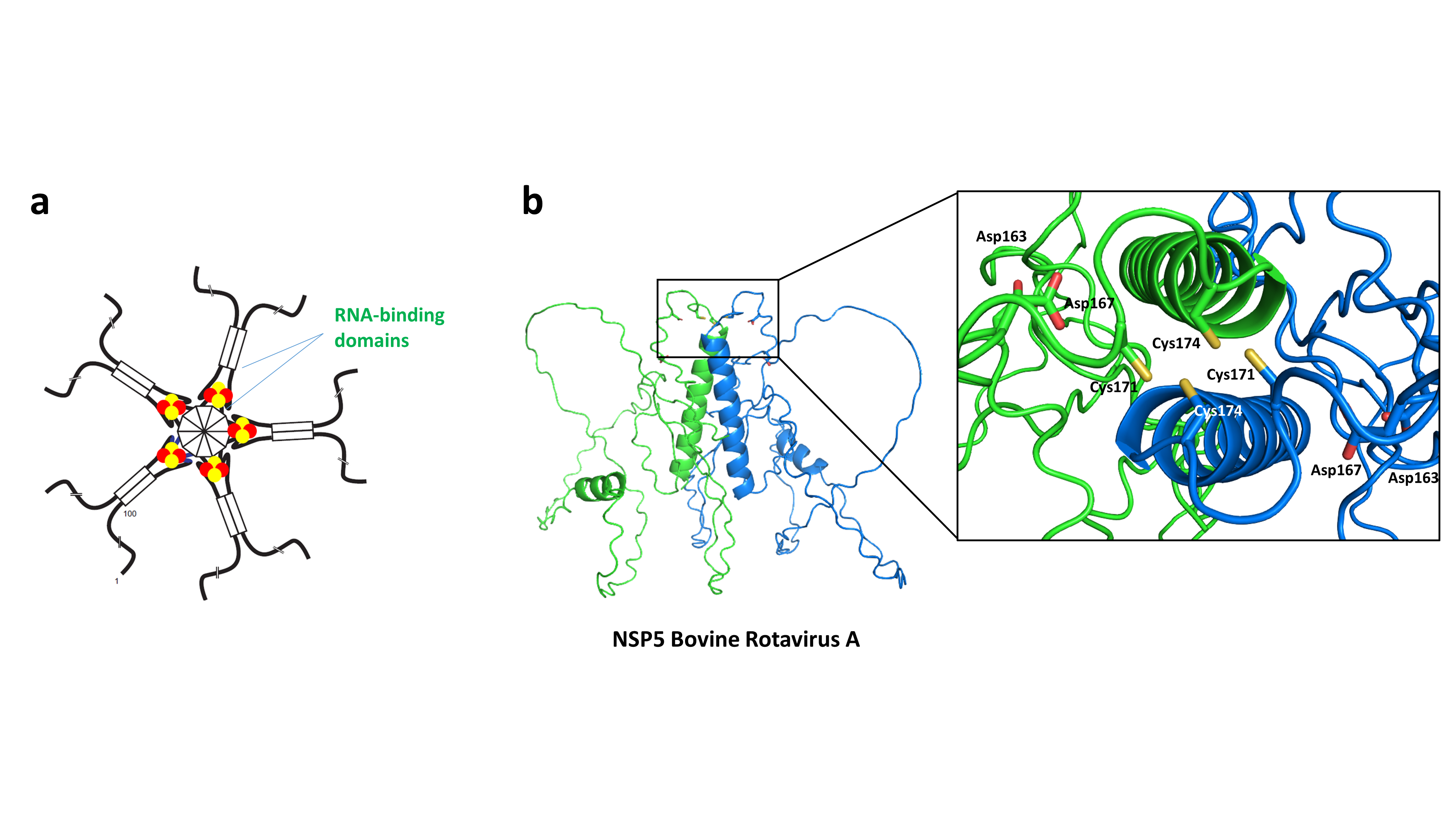
Figure 5: Structure and topology of the Fe-S cluster in NSP5
(a) Structural model of the NSP5 decamer as a pentamer of dimer. The [2Fe2S] is bound in a bridging manner at the interface of the dimer. (b) alphafold model showing a zoom on the region of cysteines 171 and 174.
OUR CURRENT EFFORTS FOCUS ON:
i) Elucidation of the functional role of Fe-S cluster in the NSP5 proteins
ii) Elucidation of the role of Fe-S cluster in other proteins