Interactions and assembly mechanisms
of proteins and Biopolymers
Topics

Structural virology
Stéphane Bressanelli
We pursue collaborations with molecular and cellular virology groups. We aim at promoting the use of structural analyses by biologists. Our principal tools, X-ray crystallography and cryo-electron microscopy, allow determination of the atomic structures of viral components. One of our major interests is in virus replication (Jupin et al., PLoS Pathog. 2017) and particularly replication complexes of RNA viruses. These complexes are encoded in virus genomes and synthesize new copies of viral genetic material inside virus-infected cells (Bressanelli, Science 2015; Jupin et al., PLos Path. 2017; Ben Ouirane et al., J Biol Chem 2019).
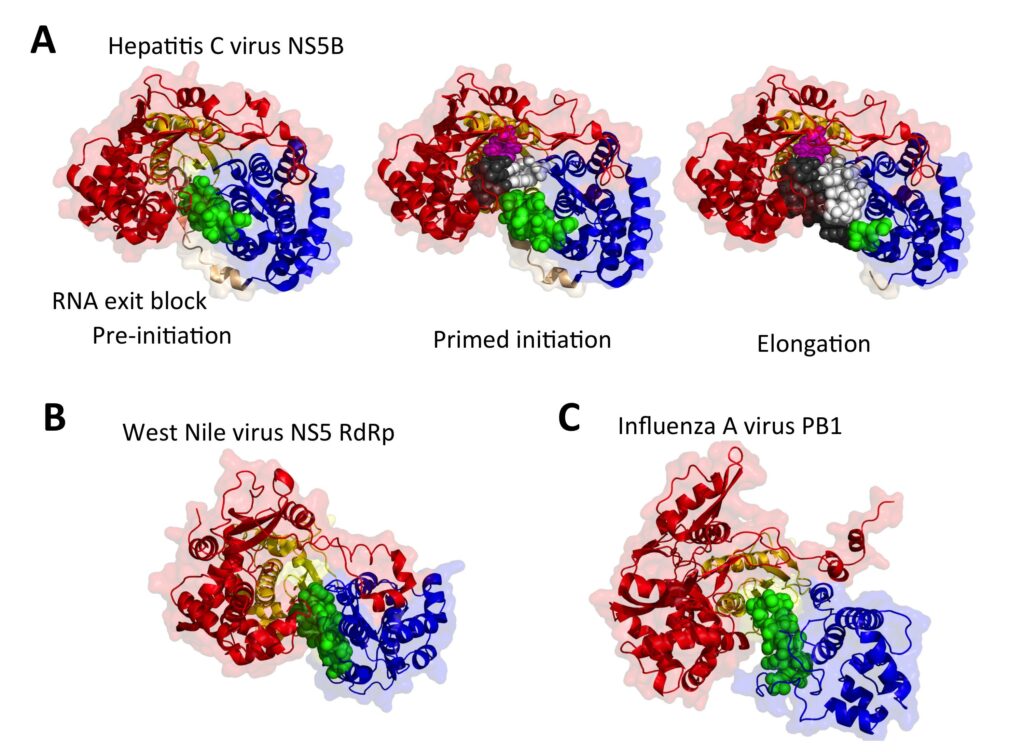
Structural virology

The proteins and enzymes of viral replication complexes are prime targets for specific antivirals that can be designed and especially improved thanks to structural analysis. Similarly, our studies on the basic physical processes underlying the self-assembly of viral capsids (Tresset et al., J. Am. Chem. Soc. 2013; Law-Hine et al., J. Phys. Chem. Lett. 2015; Lecoq et al., ChemPhysChem 2018; Chevreuil et al., Nature Commun. 2018) and the mechanisms by which enveloped viruses cross membranes to enter cells (Baquero et al., PLoS Pathogens 2015; EMBO J. 2017) are potentially connected to therapeutic applications. Our main interest however is in a fundamental understanding of the molecular mechanisms by which viruses subvert their hosts’ cellular machinery to promote their own multiplication.
Architectures of self-assembled peptides
Maïté Paternostre
This topic focuses on the mechanisms of peptide self-assembly and of the structures of the assemblies. We study the self-assembly properties of therapeutic oligopeptides with a length of 8 to 14 amino-acids. We have two goals: i) understanding and characterizing as finely as possible the physical and physico-chemical rules guiding self-assembly of these molecules and ii) find novel formulations for therapeutic peptides. This second goal is pursued within “Archi-Pex“, a multipartite lab involving three groups from three French cities, two academic groups (Franck Artzner, Institute of Physics . Rennes, and Maité Paternostre, I2BC) and one industry group (Joel Richard, IPSEN-Pharma).
Our basic studies on structures formed by two classes of natural peptidic hormones and their chemical derivatives and on the mechanisms by which those structures self-assemble have led to many first-rate publications (cf. list of publications) as well as three patents on those peptides’ formulations filed together with IPSEN-Pharma (cf. list of patents).
Structure of peptide nanotubes
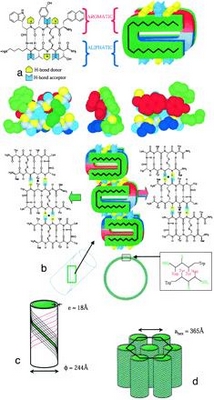
The controlled self-assembly of complex molecules into well defined hierarchical structures is a promising route for fabricating nanostructures. These nanoscale structures can be realized by naturally occurring proteins such as tobacco mosaic virus, capsid proteins, tubulin, actin, etc. Here, we report a simple alternative method based on self-assembling nanotubes formed by a synthetic therapeutic octapeptide, Lanreotide in water. We used a multidisciplinary approach involving optical and electron microscopies, vibrational spectroscopies, and small and wide angle x-ray scattering to elucidate the hierarchy of structures exhibited by this system. The results revealed the hexagonal packing of nanotubes, and high degree of monodispersity in the tube diameter (244 Å) and wall thickness (18 Å). Moreover, the diameter is tunable by suitable modifications in the molecular structure. The self-assembly of the nanotubes occurs through the association of -sheets driven by amphiphilicity and a systematic aromaticaliphatic side chain segregation. This original and simple system is a unique example for the study of complex self-assembling processes generated by de novo molecules or amyloid peptides.
Valéry et al. PNAS, 2003, 100(18), 10258–10262
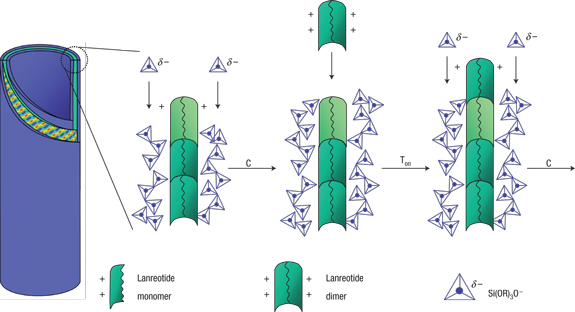
Mechanism of formation of lanreotide nanotubes
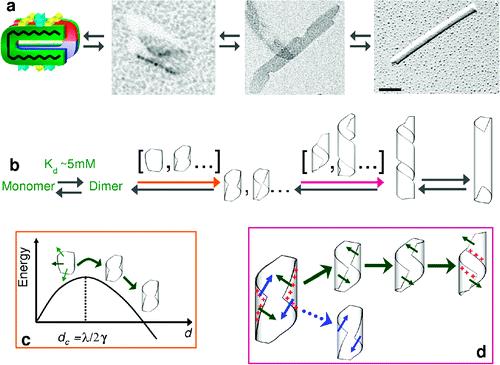
Nanofabrication by molecular self-assembly involves the design of molecules and self-assembly strategies so that shape and chemical complementarities drive the units to organize spontaneously into the desired structures. The power of self-assembly makes it the ubiquitous strategy of living organized matter and provides a powerful tool to chemists. However, a challenging issue in the self-assembly of complex supramolecular structures is to understand how kinetically efficient pathways emerge from the multitude of possible transition states and routes. Unfortunately, very few systems provide an intelligible structure and formation mechanism on which new models can be developed. Here, we elucidate the molecular and supramolecular self-assembly mechanism of synthetic octapeptide into nanotubes in equilibrium conditions. Their complex hierarchical self-assembly has recently been described at the mesoscopic level, and we show now that this system uniquely exhibits three assembly stages and three intermediates: (i) a peptide dimer is evidenced by both analytical centrifugation and NMR translational diffusion experiments; (ii) an open ribbon and (iii) an unstable helical ribbon are both visualized by transmission electron microscopy and characterized by small angle X-ray scattering. Interestingly, the structural features of two stable intermediates are related to the final nanotube organization as they set, respectively, the nanotube wall thickness and the final wall curvature radius. We propose that a specific self-assembly pathway is selected by the existence of such preorganized and stable intermediates so that a unique final molecular organization is kinetically favored. Our findings suggests that the rational design of oligopeptides can encode both molecular- and macro-scale morphological characteristics of their higher order assemblies, thus opening the way to ultrahigh resolution peptide scaffold engineering
Pouget et al, 2010, JACS, 132, 4230–4241
From single molecule to control of assembly size
Supramolecular self-assembly is an attractive pathway for bottom-up synthesis of novel nanomaterials. In particular, this approach allows the spontaneous formation of structures of well-defined shapes and monodisperse characteristic sizes. Because nanotechnology mainly relies on size-dependent physical phenomena, the control of monodispersity is required, but the possibility of tuning the size is also essential. For self-assembling systems, shape, size, and monodispersity are mainly settled by the chemical structure of the building block. Attempts to change the size notably by chemical modification usually end up with the loss of self-assembly. Here, we generated a library of 17 peptides forming nanotubes of monodisperse diameter ranging from 10 to 36 nm. A structural model taking into account close contacts explains how a modification of a few Å of a single aromatic residue induces a fourfold increase in nanotube diameter. The application of such a strategy is demonstrated by the formation of silica nanotubes of various diameters.
Tarabout et al, PNAS, 2011, 108(19), 7679–7684
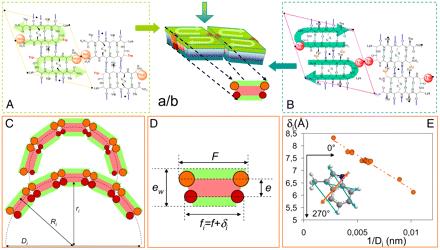
Influence of counter-ions on peptide self-assembly
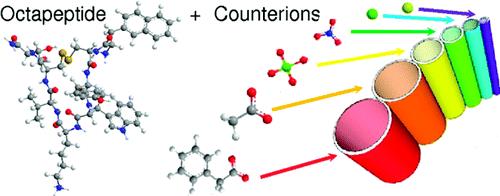
Among noncovalent forces, electrostatic ones are the strongest and possess a rather long-range action. For these reasons, charges and counterions play a prominent role in selfassembly processes in water and therefore in many biological systems. However, the complexity of the biological media often hinders a detailed understanding of all the electrostatic-related events. In this context, we have studied the role of charges and counterions in the self-assembly of lanreotide, a cationic octapeptide. This peptide spontaneously forms monodisperse nanotubes (NTs) above a critical concentration when solubilized in pure water. Free from any screening buffer, we assessed the interactions between the different peptide oligomers and counterions in solutions, above and below the critical assembly concentration. Our results provide explanations for the selection of a dimeric building block instead of a monomeric one. Indeed, the apparent charge of the dimers is lower than that of the monomers because of strong chemisorption. This phenomenon has two consequences: (i) the dimer−dimer interaction is less repulsive than the monomer−monomer one and (ii) the lowered charge of the dimeric building block weakens the electrostatic repulsion from the positively charged NT walls. Moreover, additional counterion condensation (physisorption) occurs on the NT wall. We furthermore show that the counterions interacting with the NTs play a structural role as they tune the NTs diameter. We demonstrate by a simple model that counterions adsorption sites located on the inner face of the NT walls are responsible for this size control.
Gobeaux et al., 2012, J.Am.Chem.Soc., 134 (1), pp 723–733
Use of lanreotide nanotubes as templates to form double-walled glass nanotubes.
Diatoms, shells, bones and teeth are exquisite examples of well-defined structures, arranged from nanometre to macroscopic length scale, produced by natural biomineralization using organic templates to control the growth of the inorganic phase1, 2, 3, 4, 5, 6. Although strategies mimicking Nature have partially succeeded in synthesizing human-designed bio-inorganic composite materials7, 8, 9,10, our limited understanding of fundamental mechanisms has so far kept the level of hierarchical complexity found in biological organisms out of the chemists’ reach11. In this letter, we report on the synthesis of unprecedented double-walled silica nanotubes with monodisperse diameters that self-organize into highly ordered centimetre-sized fibres. A unique synergistic growth mechanism is elucidated by the combination of light and electron microscopy, synchrotron X-ray diffuse scattering and Raman spectroscopy. Following this growth mechanism, macroscopic bundles of nanotubules result from the kinetic cross-coupling of two molecular processes: a dynamical supramolecular self-assembly and a stabilizing silica mineralization. The feedback actions between the template growth and the inorganic deposition are driven by a mutual electrostatic neutralization. This ’dynamical template’ concept can be further generalized as a rational preparation scheme for materials with well-defined multiscale architectures and also as a fundamental mechanism for growth processes in biological systems.
Pouget et al., Nature Materials, 2007, 6, 434 – 439
Interactions between proteins and nanoparticles
Yves Boulard
Our recent work (CEA biology / physics DSV/DSM collaboration) within the CEA Toxicology Program have led to the development of an original methodology allowing a better understanding of protein adsorption on silica nanoparticles. The principle lies in identifying by proteomics analyses adsorbed and non-adsorbed proteins after contact of a sulfur-35 labeled protein extract with nanoparticles. The number of proteins found in the adsorbed and non-adsorbed groups was sufficient (>30 in both cases) to 1) perform a statistical comparative analysis allowing identification of physical-chemical determinants of adsorption / non-adsorption ; 2) perform molecular dynamics simulations to assess flexibility of proteins in both groups. This approach combining physical-chemical and structural properties in both groups was then validated with commercial SiO2 nanoparticles. Our analysis shows the importance of arginine residues in adsorption, in agreement with an electrostatic mechanism of adsorption. Furthermore, the high content in aromatic residues in non-adsorbed proteins was directly correlated to their strong structuration due to numerous π–π interactions.
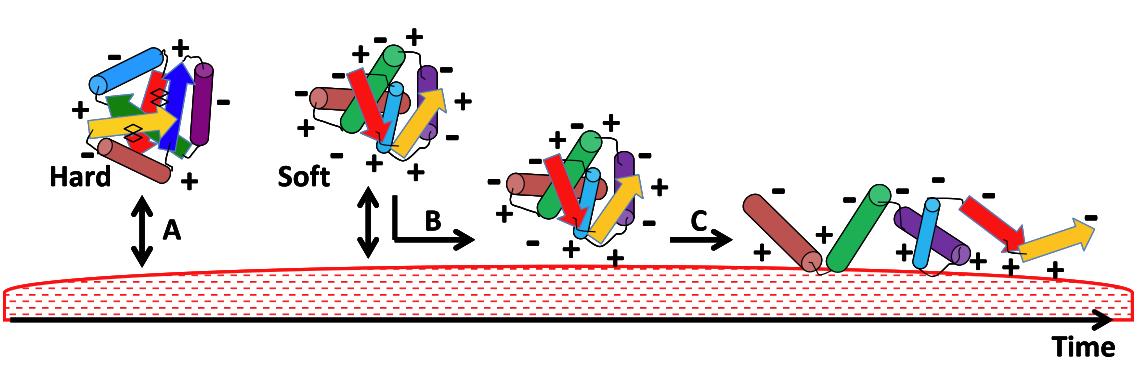
Our data show the fundamental importance of the flexible character of proteins for their adsorption (Mathé et al., 2013).
Publications
- Klein G, Devineau S, Aude J C, Boulard Y, Pasquier H, Labarre J, Pin S, Renault J P. Interferences of Silica Nanoparticles in Green Fluorescent Protein Folding Processes. Langmuir. 2016. 32: 195–202.
- Scrima N, Lepault J, Boulard Y, Pasdeloup D, Bressanelli S, Roche S. Insights into herpesvirus tegument organization from structural analyses of the 970 central residues of HSV-1 UL36 protein. Biol. Chem.2015. 290: 8820–8833.
- Tassali N, Kotera N, Boutin C, Léonce E, Boulard Y, Rousseau B, Dubost E, Taran F, Brotin T, Dutasta J-P, Berthault P. Smart detection of toxic metal ions, Pb2+ and Cd2+, using a 129Xe NMR-based sensor. Chem.2014. 86: 1783–1788.
- Dubost E, Dognon J-P, Rousseau B, Milanole G, Dugave C, Boulard Y, Léonce E, Boutin C, Berthault P. Understanding a host-guest model system through ¹²⁹Xe NMR spectroscopic experiments and theoretical studies. Chem. Int. Ed. Engl.2014. 53: 9837–9840.
- Mathé C, Devineau S, Aude J-C, Lagniel G, Chédin S, Legros V, Mathon M-H, Renault J-P, Pin S, Boulard Y, Labarre J. Structural Determinants for Protein adsorption/non-adsorption to Silica Surface. PLoS ONE. 2013. 8: e81346.
- Dubost E, Kotera N, Garcia-Argote S, Boulard Y, Léonce E, Boutin C, Berthault P, Dugave C, Rousseau B. Synthesis of a functionalizable water-soluble cryptophane-111. Lett.2013. 15: 2866–2868.
- Devineau S, Boulard Y, Labarre J. Protéines et nanoparticules, ça colle… ou pas. 2013.