Membrane Proteins
and Membrane Systems
Topics
Molecular mechanism of flippase-catalyzed transbilayer lipid transport
The plasma membrane of eukaryotic cells is made of two leaflets which display remarkably different lipid compositions, an arrangement referred to as transbilayer lipid asymmetry. Lipid asymmetry, which serves major roles in various (patho)physiological processes, is maintained by integral membrane proteins called lipid flippases. We aim to decipher the molecular mechanism of flippase-mediated transbilayer lipid transport, using biochemical and biophysical approaches.
Eukaryotic cell membranes are comprised of several thousands of different lipid species. Why would cells generate such a repertoire if lipids were only fulfilling a barrier function? As a matter of fact, lipids are essential molecules, e.g. in cell signaling, as signposts for membrane identity, or for the regulation of membrane protein activity and membrane trafficking. Beyond diversity, lipids are characterized by a non random distribution over the two leaflets of cell membranes. This so-called transbilayer lipid distribution is regulated by three kind of lipid transporters (Figure 1), the flippases (transport toward the inside), the floppases (transport toward the outside) and the scramblases (transport in both directions).
The critical role of lipid asymmetry in cell physiology is illustrated by the fact that in humans, mutations of flippases (or P4-ATPases) result in severe neurological disorders, as well as a rare liver disease called progressive familial intrahepatic cholestasis. Furthermore, select flippases have been identified as strong contributors of virulence in the pathogen fungus Cryptococcus neoformans.
Figure 1. Transmembrane lipid transporters in eukaryotic cell membranes (adapted from Montigny et al, 2016). Left, Flippases transport lipids from the exoplasmic (out) to the cytosolic (in) leaflet of membranes while floppases catalyze transport in the opposite direction. Scramblases disrupt phospholipid asymmetry by catalyzing a fast, bi-directional, energy-independent, and poorly specific transport. The arrows refer to the direction of lipid transport. Flippases and floppases use the energy of ATP to catalyze active transport. Right, Topology of P4-ATPases and their associated CDC50 subunits, with the transmembrane domain in tan and the Actuator domain (A), the Nucleotide binding domain (N) and the Phosphorylation domain (P) in yellow, red, and blue, respectively. Cdc50 with two transmembrane spans and a large exoplasmic loop in pink; predicted disulfide bridges (S-S) and glycosylation sites (green) are indicated.
In this context, we aim to decipher the molecular mechanism of flippase-catalyzed transbilayer lipid transport, and the link between membrane remodeling and vesicle biogenesis. To this end, we devised a procedure for the co-expression and purification of the S. cerevisiae Drs2/Cdc50 flippase complex in a functional state (Jacquot et al, 2012; Azouaoui et al, 2014; Azouaoui et al, 2016) and identified phosphatidylserine (PS) and phosphatidylinositol-4-phosphate (PI4P) as key to the stability of the purified complex (Azouaoui et al, 2014). This allowed us to identify a critical role for the N- and C-terminal extensions of Drs2 in auto-inhibiting the complex and the contribution of PI4P in regulating its activity (Azouaoui et al, 2017). Using this pure and functional Drs2/Cdc50 complex, we recently obtained in collaboration with the laboratory of Poul Nissen (Aarhus University, Denmark) the first high-resolution structure of a lipid flippase (Timcenko et al, 2019) (Figure 2).
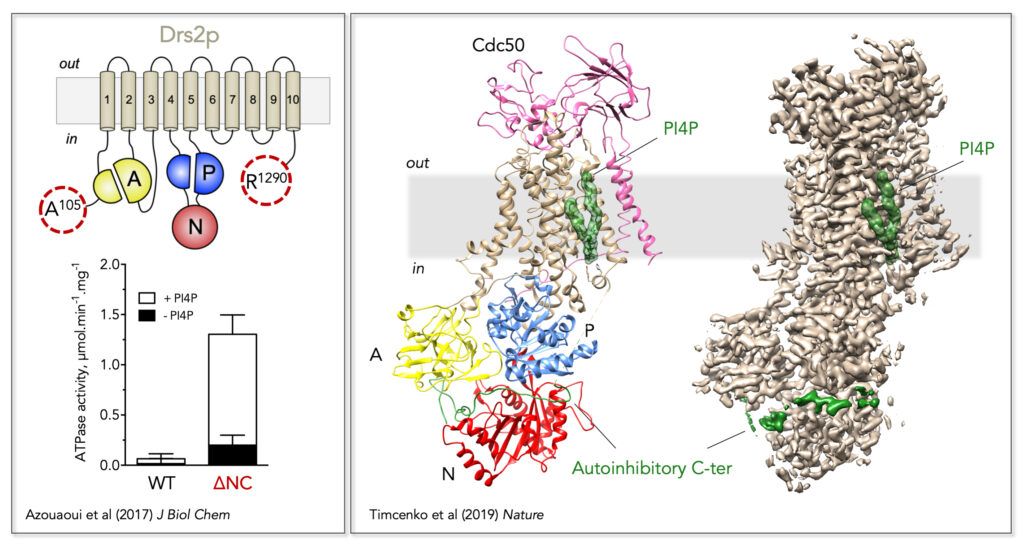
Figure 2. Regulatory mechanism of the yeast Drs2/Cdc50 phospholipid flippase. Left, Trimming of the N- and C-terminal extensions of Drs2 dramatically increases its activity, which nevertheless remains dependent on PI4P. Right, Localization of Drs2/Cdc50 regulatory elements on its three-dimensional high-resolution structure (PDB: 6ROI, EMD: 4973). The Drs2 transmembrane domain is displayed in tan, the A domain in yellow, the N domain in red, and the P domain in blue on the structural model (left). PI4P is displayed as green surface and the autoinhibitory C-terminus as a green cartoon on the structural model. Both PI4P and the C-terminus are also shown on the EM map (right).
Both in the short and the long term, we develop the following projects :
– Regulatory mechanism the Drs2/Cdc50 complex; although we know the complex is auto-inhibited by its terminal extensions, which part of the C-terminus is necessary for actual auto-inhibition remains to be elucidated. Also, what is the contribution of the N-ter in auto-regulation? Similarly, although the binding site for PI4P has been identified (Figure 2), the mechanism by which PI4P activates the flippase complex is unknown.
– In vitro reconstitution of the lipid translocation machinery using purified components. Previous studies have shown that Drs2-interacting proteins Gea2 and Arl1 activate lipid transport in vivo. We want to characterize the interactions between Drs2/Cdc50, Gea2 and Arl1 in vitro as well as the possible requirement fo specific lipids.
– Functional consequences of disease-causing mutations in ATP8B1. Mutations in the human P4-ATPase ATP8B1 have been linked to progressive familial intrahepatic cholestasis. We aim at establishing the purification of a functional ATP8B1/CDC50A complex and studying the consequences of ATP8B1 mutations on its activity.
– Biochemical study of putative flippases from C. neoformans. The P4-ATPase Apt1 is involved in C. neoformans virulence. First, is Apt1 a genuine lipid flippase? Then, we wish to screen small molecules in order to find inhibitors of the complex.
Contact person: Guillaume Lenoir

Investigation of caveolin-1 folding, assemblies and membrane interactions
In addition to transversal heterogenity, cell membranes present lateral heterogeneity due non-homogeneously distribution of proteins and lipids within the membrane plane. Transversal and lateral heterogeneity are interdependent. Portions of membranes with distinct composition from the rest of the plasma membrane are named membrane domains. These domains, depending on their composition, have different physical and chemical properties, such as thickness, elasticity, long or short chains saturated lipids, cholesterol, protein crowding, etc… and thus different functionnal roles. Caveolae are « stable » membrane domains enriched in Caveolin-1, Cavins, and specific lipids such as cholesterol. Caveolin-1 is a small monotopic membrane protein found at the plasma membrane in caveolae and non-caveolae domains as caveolin-1 oligomers. It is a key structural protein of caveolae as it possesses the intrinsic properties to curve its membrane. The complex assemblies of caveolin-1 and lipids at the plasma membrane represent a challenge for the understanding of the molecular and functionnal properties of caveolin-1.
In that context, we aim at understanding the relationship between the original amino acid sequence of Caveolin-1 and its properties of oligomerization and shaping the membrane at a molecular level. For that purpose, we have produced and characterized assemblies of caveolin-1 fragments and detergent by high resolution magnetic resonance spectroscopy and modelisation, and we are now pursuing our work using Caveolin-1β , fragments and mutants assemblies with different detergents and lipids using a range of biophysical techniques.
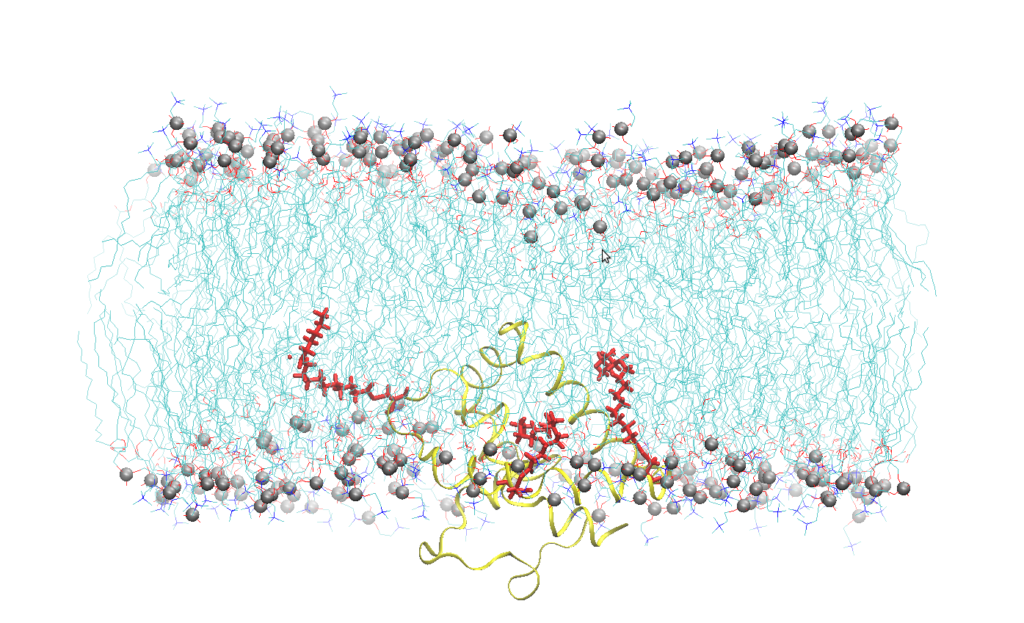
Figure 1. Snapshot of Cav-1(76-178) embedded into a SOPC bilayer after 200 ns of all atoms molecular dynamics simulation. Lipids are represented by blue lines, phosphorus atom of the lipid head groups by silver sphere, Cav-1 (76-178) by a yellow ribbon, the palmitoyl chain by red sticks. Water molecules have been omitted for the sake of clarity.
The overexpression of Caveolin-1β can produce intracellular membrane vesicles called heterologous caveolae (h-caveolae) in the host organisms. We have observed this production in Escherichia coli bacteria and in the Sf21 insect cells, and we propose to produce membrane protein-enriched vesicles using the ability of Caveolin 1β to generate membrane vesicles within the cytoplasm in various hosts. As a proof of concept, we have chosen to co-express with Caveolin-1β the human microsomal glutathione S-transferase (hMGST1) in Escherichia coli and in the Sf21 insect cells. hMGST1 belongs to the membrane-associated proteins in eicosanoid and glutathione metabolism (MAPEG) family of integral MPs and catalyze glutathione-dependent modifications of lipophilic substrates present in the lipid membranes. hMGST1 is highly abundant in liver, and displays a noticeable role in xenobiotic detoxification, including some cytotoxic drugs.
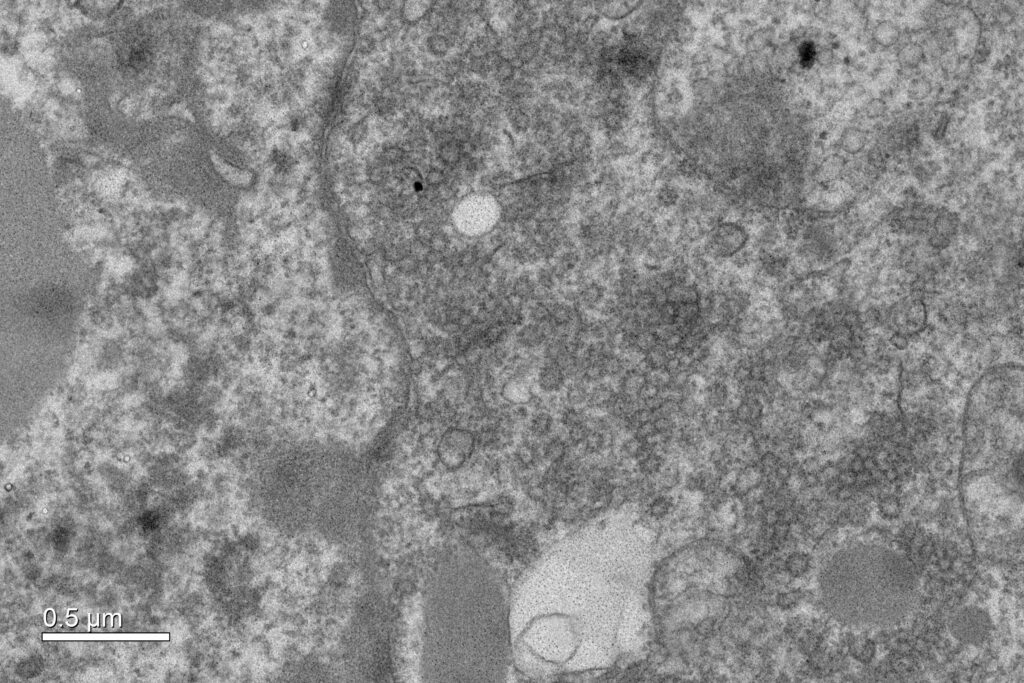
Figure 2. Cytoplasmic vesicles induced by the over-expression of Cav1β in sf21 insect cells. Electron micrograph recorded at Imagif platform with the expertise of Cynthia Gillet and Claire Boulogne.
Contact person: Nadège Jamin

Structure, function and regulation of disease-related membrane transport proteins
The physiological and therapeutical relevance of Membrane Transport Proteins (MTPs) is reflected by the fact that MTPs comprise ~15 % of the FDA-approved human targets, and that ~18 % of total FDA-approved drugs target MTPs. We study the structure-function relationships of disease-related amino acid transporters and transporters encoded by malaria parasites. Our goal is to understand their biological role and pathophysiology, as well as to find new mechanisms for pharmaceutical intervention. To this end we combine structural biology, spectroscopy and biochemical tools, including protein engineering.
1. Amino acid transporters.
Amino acid transporters are essentials players in human physiology as they participate on protein synthesis, energy metabolism, gene expression, redox balance or signal transduction pathways. One of the largest families of amino acid transporters is the L-amino acid transporter (LAT) family, where congenital mutatios of two members of this family cause two pathological disorders: cystinuria and lysinuric protein intolerance. In collaboration with Prof Manuel Palacín (IRB, Barcelona, Spain) we solved the X-ray structure of a LAT prokaryotic homolog: Adic (Kowalczyk, L et al, 2011, Figure 1), revealing for the first time an induced-fit mechanism during the first step of substrate-protein interaction. While two structures of mammalian LATs have recently appeared, prokaryotic LATs are still very valuable tools to continue deciphering the transport mechanism and regulation of their mammalian homologs. Using a directed evolution strategy (Rodriguez-Banqueri et al 2016), we have generated engineered versions of SteT (a prokaryotic paradigm of LATs) trapped in different stages along the catalytic cycle. Structure-function analyses of these mutants are expected to improve our understanding of the transport mechanism and lipid regulation of LATs.
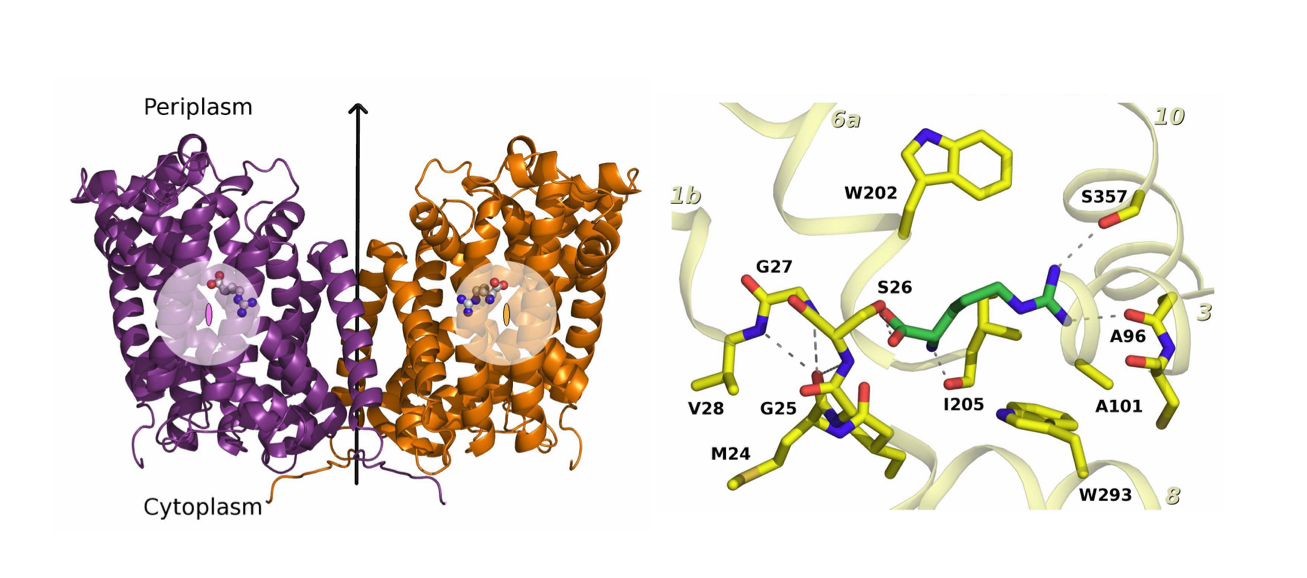
Figure 1. Structure of a mutated version of Adic bound to the substrate, Arg+. (A) The transporter forms a homodimer (in purple, protomer 1 and in orange, protomer 2). The bound substrate is depicted with a ball-and-stick model. (B) Detail of the binding site. The structure shows the delocalization of the substrate in the binding site as a consequence of removing one hydrogen bond interaction between the substrate and the transporter, providing structural evidences of a substrate’s induce fit mechanism of these transporters.
2. Membrane transporters from malaria parasites
Due to the intracellular mode of living of Plasmodium parasites, MTPs encoded by these parasites are a source of potential candidates for drug targeting, although most of them are still considered as putative. Notably, a high proportion of transporters with predicted role in lipid transport and in the maintenance of membrane lipid asymmetry are essential for the parasite; like ATP2, the Plasmodium P4-ATPase (or lipid flippase), recently identified as the possible target of two antimalarial drug candidates. Recombinant ATP2 associates with two of the three Plasmodium-encoded Cdc50 proteins (Cdc50A and Cdc50B), and functional analysis of purified ATP2/Cdc50B complex have identified possible physiological phospholipid substrates of ATP2 (Lamy et al, 2020). Our next goals include a better understanding of the catalytic activity of ATP2 and the physiological meaning of ATP2 association with each of the Cdc50 subunits, and the finding of ATP2 inhibitors. We also aim at determining the 3D atomic structure of ATP2 in complex with these Cdc50 subunits.
3. Development of new methodologies to study MTPs
MTPs are notoriously challenging proteins to produce in heterologous host. Also, their metastable nature is often an obstacle for their structural and functional characterization. We have developed new approaches and methodologies to circumvent some of these problems:
- We developed a strategy to engineer stability in a LAT transporter combining random mutagenesis, and a split-GFP complementation assay as reporter of protein expression and membrane insertion (Rodriguez-Banqueri A et al, 2012), (Rodriguez-Banqueri A et al, 2016), (Errasti-Murugarren E et al, 2017).
- We have used nanobodies (the heavy chain domains of camelide antibodies) against the GFP to purify the GFP-tagged ATP2 in complex with a Cdc50 beta-subunit, and to immobilize them in agarose beads for functional analysis (Lamy et al, 2020). In collaboration with Leandro Tabares and Sun Un, we are now aiming to extend the use of nanobodies for in vivo and in vitro structural characterization of MTPs (and complexes) using spectroscopic tools.
Lamy, A. et al. bioRxiv06.08.121152 (2020). doi:10.1101/2020.06.08.121152
Errasti-Murugarren E, Rodríguez-Banqueri A, and Vázquez-Ibar JL. Methods Mol Biol. (2017); 1586:181-195.
Rodríguez-Banqueri A et al. J Gen Physiol. (2016); 147(4): 353-68.
Rodríguez-Banqueri A. et al. Anal Biochem. (2012); 423(1): 7–14.
Kowalczyk, L et al Proc. Acad. Sci. U.S.A. (2011); 108(10): 3935-40.