Photobiology, Photosynthesis, Photocatalysis
Topics
Natural photosynthesis
Sandrine Cot, Diana Kirilovsky, Anja Krieger-Liszkay, Thanh-Lan Lai, Lucas Leverne, Marine Messant, Pavel Müller, Annamaria Quaranta, Pierre Sétif, Corinne Rivasseau, Adjélé Wilson
Light is essential for photosynthetic organisms which convert solar energy into chemical energy necessary for the synthesis of sugars and for O2 evolution. However, light becomes dangerous when the energy arriving at the reaction centers of the photosynthetic apparatus exceeds the energy consumption by multiple cellular processes. In this case, the photosynthetic electron transport chain becomes reduced and reactive oxygen species (ROS) leading to severe damage of the cells are accumulated, especially at the level of the photosynthetic apparatus. This occurs at high light intensities but other stress conditions, like drought or nutrient starvation can provoke this disequilibrium even at lower irradiance.
Photosynthetic organisms have to protect themselves when the light intensities are higher than needed for photosynthesis and under other abiotic stress conditions. We are studying these protection mechanisms with a special focus on (1) the regulation of the photosynthetic electron transport chain with a special focus on photosystem II, the redox state of the plastoquinone pool and the plastid terminal oxidase PTOX, and (2) the regulation and mechanism of heat dissipation (non-photochemical quenching) in cyanobacteria and plants, and (3) the extreme adaptation response of Coccomyxa actinabiotis sp. nov that was isolated from a cooling pool of a nuclear reactor.
Regulation mechanisms of photosynthetic electron transport
a) Redox regulation of photosystem II
We have demonstrated that the energetics of photosystem II depend on the activity of the water-splitting complex [Mn4CaO5] cluster at its donor side and on the molecular environment of the primary quinone acceptor QA at its acceptor side. At present we investigate the importance of the assembly of the cluster and of bicarbonate at the acceptor side of photosystem II for tuning the midpoint potential of QA and for photoprotection against the ROS singlet oxygen. We aim to elucidate the role of photorespiratory metabolites in replacing bicarbonate to demonstrate the coordination between primary reactions, photosynthetic metabolites and protection against photo-oxidative stress.
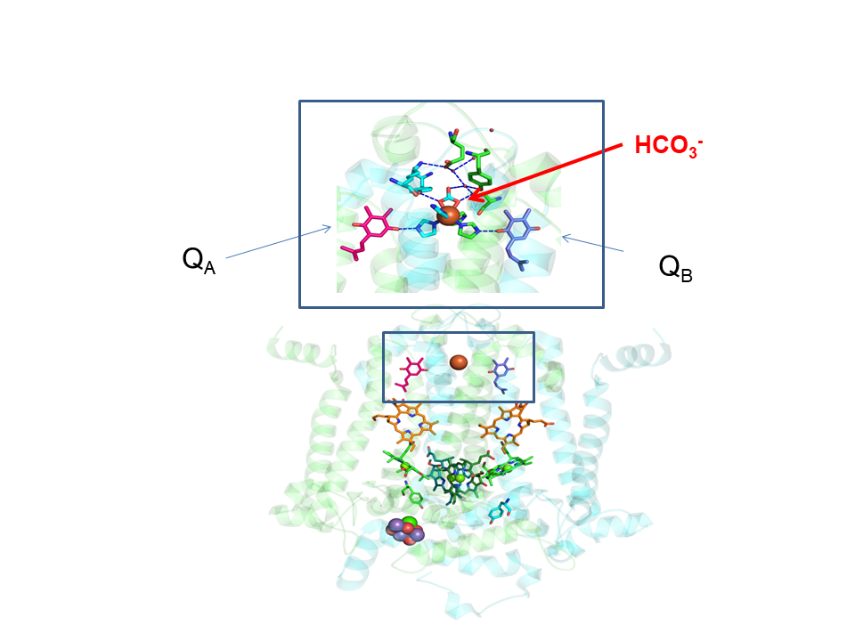
Structure of Photosystem II with a zoom on the acceptor side with the quinone acceptors QA and QB linked by the non-heme iron
b) Alternative electron transport: dynamics and molecular characterization of the plastid terminal oxidase PTOX.
PTOX oxidises plastoquinol by transferring electrons to O2 and generating water. PTOX activity may be important to set the redox poise of the photosynthetic electron transport chain under stress condition. The amount of PTOX increases under conditions of oxidative stress. We have biochemically characterized PTOX and established a dynamic localization of PTOX within the chloroplast. At present, we are studying the physiological role of PTOX, its interaction partners and its catalytic activity on a molecular level.
Overexpression of PTOX in tobacco:
Left: quantum yield of photosystem II dark blue: higher value than light blue). Right: non-photochemical quenching (light blue: higher value than green)
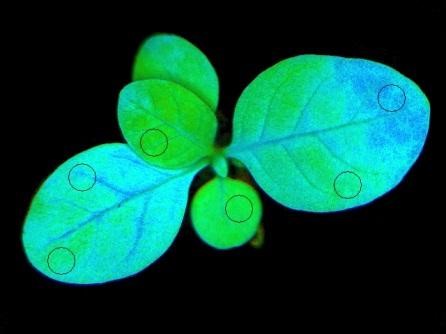
Non-photochemical quenching in cyanobacteria and higher plants
We have demonstrated that the photoactivation of a soluble carotenoid protein, the Orange Carotenoid Protein (OCP), is essential for the triggering of a photoprotective mechanism that decreases the energy arriving to the photochemical centers. The OCP is part of the family of photoactive proteins, which are essentials elements to detect environmental changes. It is the first photoactive protein containing a carotenoid as active chromophore and having an original and unique mechanism of photoactivation. We largely contributed to the understanding of OCP activity and OCP-related-photoprotective mechanism. We also studied other carotenoid proteins (HCP, CTDHs) related to the OCP involved in photoprotection and in a new protein-protein carotenoid transfer mechanism. Nowadays, we are investigating the OCP photocycle mechanism as well as OCP interaction with the phycobilisome, the cyanobacterial antenna.
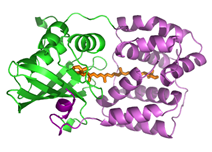
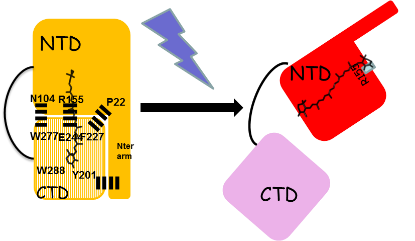
In darkness, the OCP is orange and has a compact structure.
Under strong light, it is red with an open structure in which the N-terminal (NTD) and C-terminal (CTD) domains are separated and the carotenoid is in the NTD.
Resistance to photo-oxidative stress in microalgae living in nuclear pools
Water pools in nuclear facilities such as those used to store spent fuels constitute extreme environments for life. However, green microalgae have been found living in there, withstanding radiative, nutritive and metallic stress. Our research aims at elucidating their specific properties of radioresistance, detoxification and accumulation of metals. We particularly focus on the remarkable stability of their photosynthetic apparatus and on the mechanisms implemented to counter oxidative stress generated by radiations and repair DNA damages. Fundamental knowledge of their survival strategies will allow to develop innovative applications for the bioremediation of polluted effluents, the bioextraction of strategic metallic elements, and for space biotechnologies.
Selected Publications
Wilson A., Muzzopappa F., Kirilovsky D. (2022) Elucidation of the essential amino acids involved in the binding of the cyanobacterial Orange Carotenoid Protein to the phycobilisome, Biochimica et Biophysica Acta (BBA) – Bioenergetics, Volume 1863,
https://doi.org/10.1016/j.bbabio.2021.148504.
Muzzopappa F, Wilson A, Kirilovsky D (2019) Interdomain interactions reveal the molecular evolution of the Orange Carotenoid Protein. Nat Plants 5, 1076–1086 http://doi.org/10.1038/s41477-019-0514-9
Petit, P.C.M.; Pible, O.; Eesbeeck, V.V.; Alban, C.; Steinmetz, G.; Mysara, M.; Monsieurs, P.; Armengaud, J.; Rivasseau, C. (2020) Direct Meta-Analyses Reveal Unexpected Microbial Life in the Highly Radioactive Water of an Operating Nuclear Reactor Core. Microorganisms, 8, 1857
Rivasseau C., Farhi E., Compagnon E., de Gouvion Saint Cyr D., van Lis R., Falconet D., Kuntz M., Atteia A., Couté A. (2016) Coccomyxa actinabiotis sp. nov. (Trebouxiophyceae, Chlorophyta), a new green microalga living in the spent fuel cooling pool of a nuclear reactor. J. Phycol. 52: 689-703
Leverne L, Roach T, Perreau F, Maignan F, Krieger-Liszkay A (2023) Increased drought resistance in state transition mutants is linked to modified plastoquinone pool redox state. Plant Cell Environ. 46:3737-3747. doi: 10.1111/pce.14695
Messant M, Hani U, Lai TL, Wilson A, Shimakawa G, Krieger-Liszkay A (2023) Plastid terminal oxidase (PTOX) protects photosystem I and not photosystem II against photoinhibition in Arabidopsis thaliana and Marchantia polymorpha. Plant J. (in press). doi: 10.1111/tpj.16520.
Messant M, Timm S, Fantuzzi A, Weckwerth W, Bauwe H, Rutherford AW, Krieger-Liszkay A (2018) Glycolate Induces Redox Tuning Of Photosystem II in Vivo: Study of a Photorespiration Mutant. Plant Physiol 177:1277-1285. doi: 10.1104/pp.18.00341
Techniques:
molecular biology, biochemistry, chlorophyll and NAD(P)H fluorescence, thermoluminescence, EPR, transient absorption spectroscopy
Photoactive proteins
Pavel Müller, Annamaria Quaranta, Adjelé Wilson, Philipp Gotico,
(Sun)light is not merely a source of heat and energy for photosynthesis. Earth-dwelling organisms have been using light in multifarious other ways than photosynthesis: to reverse UV-induced DNA damage (DNA photorepair), to convert one organic compound into another (photocatalysis), to regulate their growth and activity (photomorphogenesis, photoreception, synchronization of the circadian rhythms), to communicate, to attract a prey to eat or a partner to mate with (photo- and bio-luminescence) and most likely also to perceive the Earth’s magnetic field and to use it for orientation on the long journeys across the globe (photomagnetoreception). Behind all these processes are photoenzymes and photoactive proteins – fascinating light-harnessing molecular machines that have become increasingly sophisticated over the long course of evolution of life on our planet and without which life as we know it could never exist.
Main subjects of interest
Techniques:
Time-resolved optical spectroscopy (transient absorption, fluorescence).
DNA-repairing Enzymes Photolyases and Photoreceptors Cryptochromes
Ubiquitous DNA photolyases (PLs) and blue light receptors cryptochromes (CRYs) constitute a superfamily of evolutionarily related and structurally similar flavoproteins, which in the case of photolyases have specialized in photorepair of UV-induced DNA lesions (cyclobutane pyrimidine dimers – CPDs, and so-called 6-4 photoproducts), and in the case of CRYs function as triggers of light signalling cascades regulating photomorphogenesis of plants and entrainment of circadian rhythms in both plants and animals. CRYs are also suspected to act as photo-magnetoreceptors in migratory species.
PLs undergo two types of photoreactions: 1) photoactivation (reduction of FAD/FADH· to FADH·/FADH– via an electron transfer (ET) through a conserved chain of typically three Trp residues) and 2) DNA photorepair (catalytic C-C bond splitting in DNA lesions, which requires a fully reduced flavin FADH–). CRYs typically undergo only the former reaction (photoactivation), which in their case triggers conformational changes in their C-terminal domains, which in turn lead to interaction with other chemical partners and hence trigger signaling. Nevertheless, several examples of so-called ‘dual’ proteins have been reported, which seem to be capable of both light signaling and DNA repair.
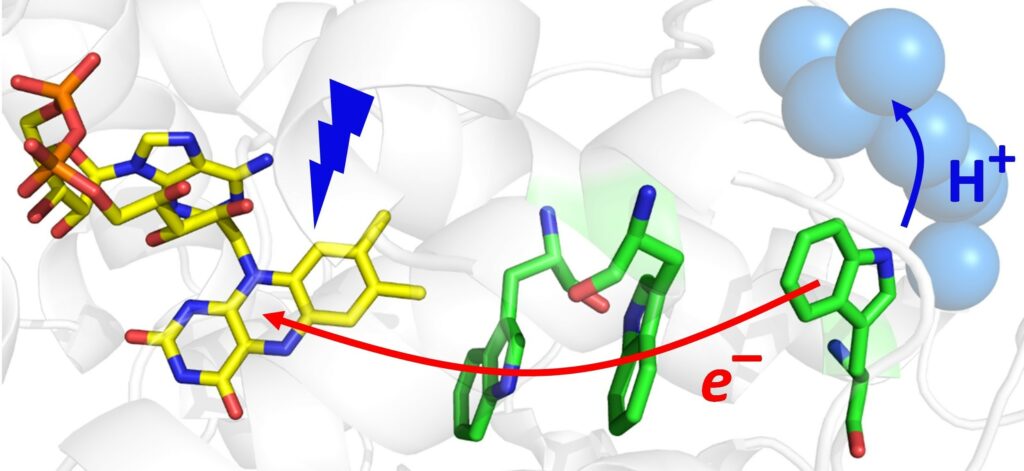
Photoactivation of cryptochromes and photolyases
In 2000, former PPP team member Klaus Brettel and collaborators established the very existence and functionality of the Trp triad and, 15 years later, Pavel Müller discovered that in certain proteins, namely in animal 6-4 PLs and animal CRYs, these ET chains involve an additional 4th Trp residue. We have predicted and later confirmed that this 4th Trp is indeed indispensable for an efficient photoactivation and thereby also (indirectly) for the photorepair of 6-4 photoproducts in the living cells. Our other significant contributions to the field include: resolution of kinetics of CPD splitting by a cyanobacterial photolyase (this one-photon process was shown to occur in 260 ps, in competition with 350 ps futile charge recombination; i.e., with >55% quantum yield), demonstration that (contrary to CPD repair) the photorepair of 6-4 photoproducts requires two photons, or that the photoactivation of plant cryptochromes is strikingly enhanced when ATP (or an ATP-like molecule) is bound in a binding pocket close to the FAD cofactor (equivalent to the DNA-binding pocket of photolyases).
Lately, we have contributed to a comprehensive time-resolved serial femtosecond X ray crystallographic (TR-SFX) study of CPD repair by a type II photolyase, visualizing the whole photorepair process on timescales ranging from picoseconds (electron transfer), through nanoseconds (DNA repair) up to microseconds (active site recovery and DNA reannealing).
Selected Publications
V. Thiagarajan, M. Byrdin, A.P.M. Eker, P. Müller & K. Brettel. Kinetics of cyclobutane thymine dimer splitting by DNA photolyase directly monitored in the UV. PNAS 2011, 108, 9402-7.
P. Müller, J.P. Bouly, K. Hitomi, V. Balland, E.D. Getzoff, T. Ritz & K. Brettel. ATP Binding Turns Plant Cryptochrome into an Efficient Natural Photoswitch. Sci. Rep. 2014, 4, 5175.
P. Müller., J. Yamamoto, R. Martin, S. Iwai & K. Brettel. Discovery and functional analysis of a 4th electron-transferring tryptophan conserved exclusively in animal cryptochromes and (6-4) photolyases. Chem. Commun. 2015, 51, 15502-5.
J. Yamamoto, K. Shimizu, T. Kanda, Y. Hosokawa, S. Iwai, P. Plaza & P. Müller. Loss of Fourth Electron-Transferring Tryptophan in Animal (6−4) Photolyase Impairs DNA Repair Activity in Bacterial Cells. Biochemistry 2017, 56, 5356-64.
P. Müller, E. Ignatz, S. Kiontke, K. Brettel & L.O. Essen. Sub-nanosecond tryptophan radical deprotonation mediated by a protein-bound water cluster in class II DNA photolyases. Chem. Sci. 2018, 9, 1200-12.
F. Lacombat, A. Espagne, N. Dozova, P. Plaza, P. Müller, K. Brettel, S. Franz-Badur, L.O. Essen. Ultrafast Oxidation of a Tyrosine by Proton-Coupled Electron Transfer Promotes Light Activation of an Animal-like Cryptochrome. J. Am. Chem. Soc. 2019, 141, 13394-409.
Y. Hosokawa, P. Müller, H. Kitoh-Nishioka, S. Iwai, J. Yamamoto. Limited solvation of an electron donating tryptophan stabilizes a photoinduced charge-separated state in plant (6-4) photolyase. Sci. Rep. 2022, 12, 5084.
K. Brettel, P. Müller, J. Yamamoto. Kinetics of Electron Returns in Successive Two-Photon DNA Repair by (6-4) Photolyase. ACS Catal. 2022, 12, 3041.
M. Maestre-Reyna et al., Visualizing the DNA repair process by a photolyase at atomic resolution. Science 2023, 382, eadd7795.
Fatty Acid Photodecarboxylase
The recently discovered photoenzyme Fatty Acid Photodecarboxylase (FAP) catalyzes the conversion of fatty acids to hydrocarbons, which is a process that bears great promise for the production of biofuels of non-fossil origin. However, how the enzyme accomplishes its function at the molecular level has not been completely clarified… until lately.
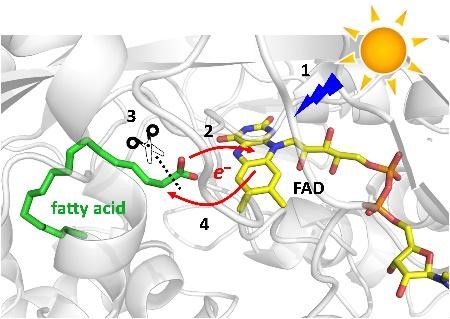
We had the privilege to be the first to study the mechanism of this photoenzyme before its discovery by the team of F. Beisson (BIAM/CEA Cadarache, France) was revealed to the public in 2017. We have established that the reaction is initiated by light-induced ET from the fatty acid carboxylate to the photoexcited FAD cofactor (in ~300 ps). This results in a C-C bond splitting and formation of CO2 and an alkyl radical. Upon back ET from FAD·– to the alkyl radical (in ~100 ns), which we suggested should be coupled to a proton transfer (PT) from an amino acid residue or a water molecule present at the active site, the oxidized state of FAD is restored. Interestingly, the spectrum of reoxidized FAD is transiently red-shifted by almost 20 nm with respect to the dark ground state (467 nm) and this shift persists for ~3 ms. We proposed that the spectral shift could be due to a change in charge of the (then unknown) proton donor XH and that the ~3 ms phase likely corresponded to the re-protonation of X–.
In a thorough follow-up study involving a large international consortium and a panoply of complementary experimental and computational techniques, we have established that the primary proton donor to the alkyl is a water molecule present at the active site. The resulting OH– then either immediately abstracts a proton from a nearby arginine residue (present at the binding site and stabilizing the substrate prior to its decarboxylation) or attacks the formed CO2 to yield bicarbonate HCO3–. The time-resolved IR experiment carried out by our collaborators at the Ecole Polytechnique in Palaiseau revealed that the majority (~75%) of the signal corresponding to the CO2 formed within the ~300 ps disappears within the subsequent ~100 ns kinetic phase at ambient temperature. The following low-temperature IR and X-ray crystallographic experiments performed in Cadarache, cryo-trapping a state with the red-shifted intermediate, provided evidence for the formation of bicarbonate.
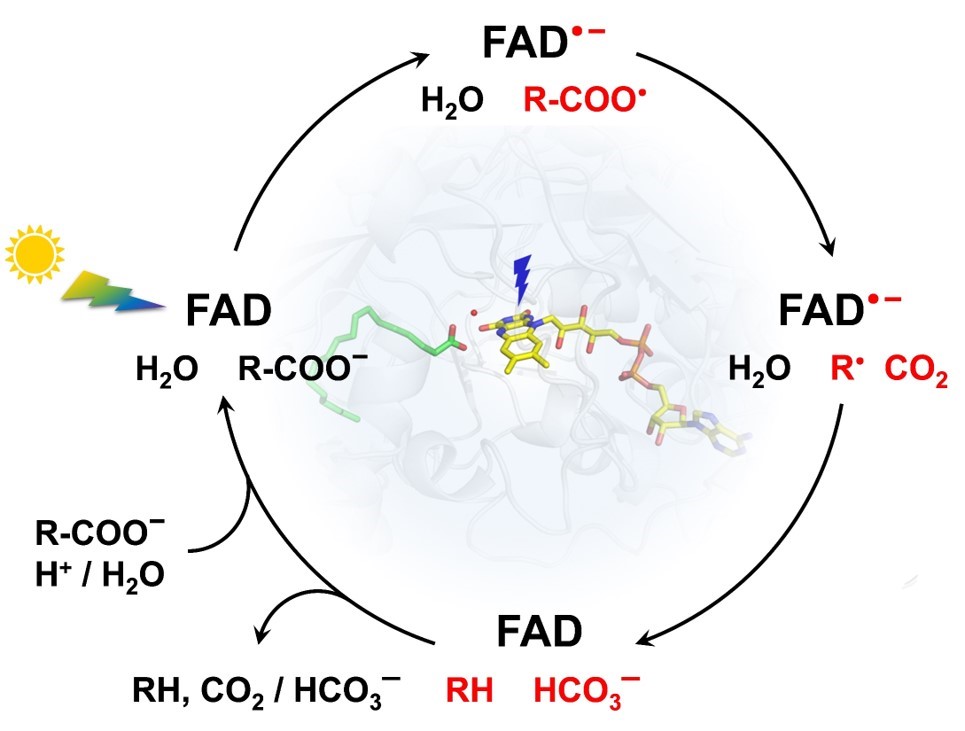
A simplified scheme of decarboxylation of fatty acids by FAP
A TR-SFX experiment, performed by our colleagues from Grenoble and Heidelberg, has not only confirmed the disappearance of the water/OH– electron density (upon its reaction with CO2), but it has also shown, that the oxidized FAD cofactor in FAP is not planar but bent already prior to its photoexcitation/photoreduction. This atypical conformation of oxidized FAD is likely responsible for a shift of the blue absorption band of the dark ground state FAD (with a maximum typically around 445-450 nm in other flavoproteins to 467 nm in FAP) towards the so-called ‘green gap’ in the absorption spectrum of chlorophylls, which should increase the net light-harvesting capabilities of FAP in vivo.
Photodecarboxylation by FAP seems to be most efficient for long chain (C14-C18) fatty acids but we have recently shown that octanoic acid (C8) is also decarboxylated very efficiently by the wild-type enzyme – due to an autocatalytic effect of the C7 hydrocarbon product, which mimics the missing part of the long chain in the substrate-binding tunnel. We are currently trying to optimize both the enzyme and/or the reaction conditions for an efficient decarboxylation of other short fatty acids, which would yield gaseous and liquid hydrocarbons readily usable as green combustible fuels with near-zero carbon footprint.
Selected Publications
D. Sorigué, et al. An algal photoenzyme converts fatty acids to hydrocarbons. Science 2017, 357, 903-7.
D. Sorigué, et al. Mechanism and dynamics of fatty acid photodecarboxylase. Science 2021, xx, xx-xx (accepted).
P.P. Samire et al. Autocatalytic Effect Boosts the Production of Medium-Chain Hydrocarbons by Fatty Acid Photodecarboxylase. Science Adv. 2023, 9 (13), eadg3881.
Fluorescent proteins
As genetically-encoded markers, fluorescent proteins (FPs) from the green fluorescent protein (GFP) family are ubiquitous tools in biological imaging. FPs exhibit a variety of photochemical reactions (photoinduced PT/ET, cis/trans photoisomerization, covalent chromophore/residue modifications…), the mechanisms of which are only partially understood.

Since the isolation of Green Fluorescent Protein (GFP) from jellyfish in the 1960’s,
proteins fluorescing throughout the visible spectrum have been engineered.
We study the rich photochemistry of FPs in collaboration with the former members of our team, Agathe Espagne (now ENS Paris) and Martin Byrdin (now IBS Grenoble). We are particularly interested in the mechanisms, dynamics and yields of the processes occurring in FPs, the knowledge of which should help to design tailor-made pulse sequences for sophisticated imaging techniques, enabling a selective activation of a single protein of choice in complex mixtures of spectrally overlapping fluorescent probes.
Selected Publications
M. Byrdin, C. Duan, D. Bourgeois & K. Brettel. A Long-Lived Triplet State Is the Entrance Gateway to Oxidative Photochemistry in Green Fluorescent Proteins. J. Am. Chem. Soc. 2018, 140, 2897-905.
K. Brettel, A. Espagne, L. Jullien, T. Le Saux, L. Ludvíková, P. Müller, L. Robert. Method for enhancing the photoresistance of a fluorescent species and fluorescence microscopy system suitable for implementing said method. PSL, CNRS, Sorbonne Univ., Univ. Paris-Saclay, INRAE, CEA & AgroParisTech, 2021, Patent EP21305834.
E. Renouard, M. Nowinska, F. Lacombat, P. Plaza, P. Müller, A. Espagne. Multiscale Transient Absorption Study of the Fluorescent Protein Dreiklang and Two Point Variants Provides Insight into Photoswitching and Nonproductive Reaction Pathways. J. Phys. Chem. Lett. 2023, 14, 6477-6485.
Turning naturally non-photoactive proteins photoactivatable
In order to study ET/PT reactions or localize a particular protein in a biological system, it is not only convenient but often also necessary to trigger reactions in a significant fraction of the present molecules simultaneously, ideally in a selective and not just temporally but also spatially controlled manner. This is best accomplished by initiating the reactions by light – either by using so-called “caged” compounds (small biologically active molecules or ions temporarily deactivated or trapped by photoremovable protecting groups), or by a direct excitation of the chromophore of an organic macromolecule (typically a cofactor of a photoactive protein).
The systems that we have studied to these days are mostly naturally photoactive proteins (or their mutants). However, most proteins are not naturally photoactive and, in order to localize them in the cell and/or study the mechanism of their function, they must be made photoactive or photoactivatable artificially. In vivo, this is increasingly achieved by attaching a photoactive protein (or a construct based on it) to the non-photoactive one already at the level of the genome, which then encodes their joint production (e.g. FPs in fluorescence imaging or CRY-based constructs in optogenetics). In vitro, light-triggered electron or hole injectors can be attached to an isolated protein (or a peptide) by a chemical reaction. In the past, we have studied the photoreduction of the ferredoxin / ferredoxin NADP+ reductase complex by a linked ruthenium polypyridyl chromophore.
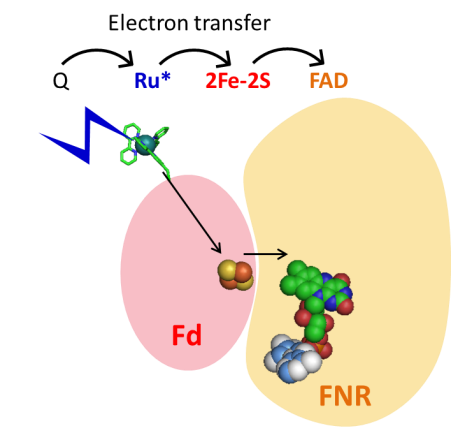
Reduction of the FAD cofactor of Ferredoxin NADP+ reductase (FNR) by ferredoxin (Fd) induced by light excitation of
and ET from a ruthenium polypyridyl complex (Ru).
Ru is re reduced by ET from a sacrificial electron donor (Q), e.g. ascorbate or ferrocyanide.
Similar approach is to be applied within the starting project in collaboration with the teams of Laura Baciou, Fabien Cailliez and Aurélien de la Lande (ICP, Univ. Paris-Saclay) and Marc Baaden (LBT, IBPC Paris), addressing the mechanism of function of NADPH oxidases by combining computational and experimental methods. We intend to use a chemically modified NADPH oxidase with a ruthenium complex grafted near the active site, which should inject electrons to the system upon photoexcitation by a short laser pulse. Using time-resolved spectroscopic techniques, we hope to be able to resolve the individual ET steps from the FAD chromophore via the two inter-membrane Hemes down to oxygen molecules (forming superoxide, which destroys pathogens in vivo).
Selected Publications
A. Quaranta, B. Lagoutte, J. Frey, P. Sétif, Photoreduction of the ferredoxin/ferredoxin–NADP+-reductase complex by a linked ruthenium polypyridyl chromophore.
J. Photochem. Photobiol B: Biology, 2016, 160, 347-354.
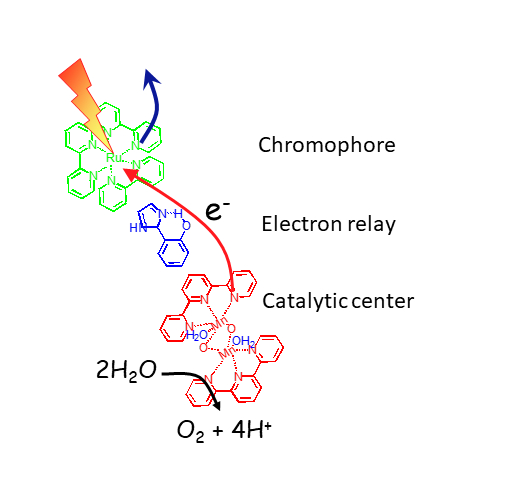
Bio-inspired photocatalysis
Annamaria Quaranta, Winfried Leibl, Philipp Gotico
Among the most important fundamental questions in chemical research one is how we can convert abundant stable molecules such as H2O, CO2, O2 and N2 into an energetic carrier and provide valuable chemical reactants for industrial usage to mitigate the environmental burden connected with the use of fossil resources. Nature provides us with main clues on how to do so for example in photosynthesis, where sunlight is used to drive multi-electron catalysis, in particular the four-electron and four-proton water oxidation.
Our team is engaged in a strong collaboration with the team of Prof. Ally Aukauloo on bio-inspired design of cheap, robust, and efficient molecular catalysts for artificial photosynthesis. The advantage of a molecular approach is the possibility of virtually unlimited rational variation of the molecules synthesized to optimize their properties. However, synthetic chemistry must be guided by functional characterization to identify unwanted side reactions, rate-limiting reaction steps and degradation pathways, which represent bottlenecks for performance. Therefore, real understanding of the photophysical properties and catalytic mechanism of all developed complexes is an important goal for us.
Main challenges currently addressed
Techniques
Time-resolved spectroscopy / (Photo)Electrochemistry / Photocatalysis
Controlling light-induced electron transfer
In natural and artificial photosynthetic systems light absorption by a chromophore leads first to formation of an excited state with an electron lifted from the highest occupied molecular orbital to the lowest unoccupied molecular orbital. Such excited states are both good oxidants and good reductants which can engage in electron transfer (ET) reactions to suitable electron donors or acceptors.
The photoredox behaviour of chromophores can be used to activate catalysts for oxidative and reductive catalytic reactions. To achieve high efficiency the rate and the directionality of these ET steps have to be controlled. This is especially important in the case of multi-electronic catalytic processes like water oxidation, or proton, carbon dioxide, and dioxygen reduction. Electron relay functions, control of distance, and proton-coupled ET are important features investigated by us in enzymes and supramolecular constructs.
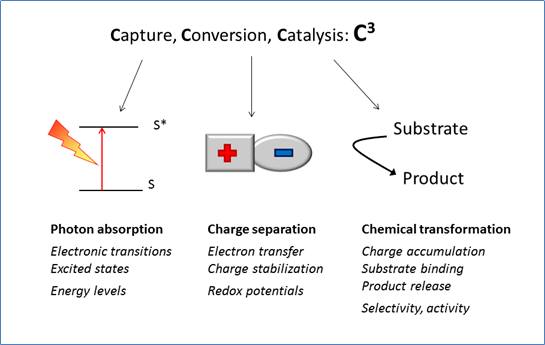
The three basic functions of photocatalytic systems and related processes and properties
We have investigated the role of the bridge in intramolecular electron transfer reactions occurring in molecular dyads and photocatalytic systems. Inspired by PSII, we have designed and characterized molecular complexes containing a hydrogen-bonded imidazole-phenol moiety to mimic the function of the TyrZ-His190 cofactors in natural photosynthesis. In the different systems investigated, phenol oxidation occurred through a PCET mechanism involving the hydrogen bonded imidazole acting as a (transient) base, the rate depending on the driving force for the electron transfer reaction.
Another connecting unit resulting from click chemistry synthetic procedures is the triazole function. We have shown in a series of dyads that such a bridge allows efficient bi-directional electron flow between two connected moieties according to their reduction potentials.
Besides the efficient conversion of the excited state into a charge-separated state, another challenging task is the ability to store the number of charges required to advance through the photocatalytic cycle. This is not a trivial task! In order to unveil and minimize unfruitful reactions (energy transfer/charge recombination) we employ a double laser excitation set-up to kinetically track each step in the charge accumulation process in view of optimizing the charges flow toward the catalytic site. We investigate supramolecular as well as multicomponent systems. Although the striking advantage of linked systems is the possibility to assure high rates for electron transfer, the direct consequence is that the back reactions are also fast. For this reason frequently higher yields of charge accumulation are observed in multicomponent systems once the concentrations have been optimized.
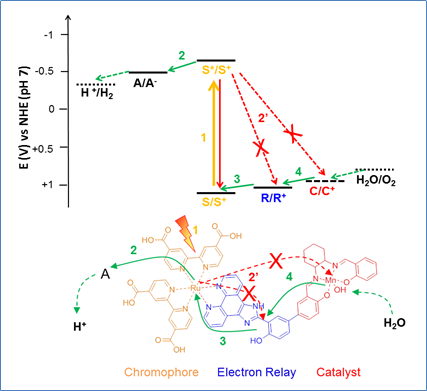
Simplified energetic scheme (top) and sequence of light-induced electron transfer reactions in a molecular photocatalyst for oxidation. Productive charge transfers are indicated by green arrows, non-productive ones by red arrows. Possible back reactions from the reduced acceptor (A–) to oxidized species (S+, R+, C+) are omitted for clarity and because in most studies in solution these back reactions are suppressed by the use of irreversible electron acceptors.
Selected Publications
Water molecules gating a photoinduced one electron two protons transfer in a Tyr/His model of Photosystem II. G. Charalambidis, S. Das, A. Trapali, A. Quaranta, M. Orio, Z. Halime, P. Fertey, R. Guillot, A. Coutsolelos, A. Leibl, A. Aukauloo, M. Sircoglou, Angew Chemie Int Ed, 2018, 57, 9013-9017.
Snapshots of Light Induced Accumulation of Two Charges on Methylviologen using a Sequential Nanosecond Pump–Pump Photoexcitation; T.-T. Tran, M.-H. Ha-Thi, T. Pino, A. Quaranta, C. Lefumeux, W. Leibl and A. Aukauloo,, J. Phys. Chem. Lett., 2018, 9, 1086–1091.
Time-Resolved Interception of Multiple-Charge Accumulation in a Sensitizer–Acceptor Dyad. S. Mendes marinho, M.-H. Ha-Thi, V.-T. Pham, A. Quaranta, T. Pino, C. Lefumeux, T. Chamaillé, W. Leibl, A. Aukauloo, Angewandte Chemie International Edition, 2017, 11, 15936–15940.
Tracking charge accumulation in a functional triazole-linked Ru-Re dyad towards photocatalytic CO2 reduction. P. Gotico, T.T. Tran, A. Baron, B. Vauzeilles, C. Lefumeux, M.H. Ha-Thi, T. Pino, Z. Halime, A. Quaranta, W. Leibl, A. Aukauloo, ChemPhotoChem 2021, 5, 654-664.
Proton-controlled action of an imidazole as electron relay in a photoredox triad. P. Gotico, C. Herrero, S. Protti, A. Quaranta, S. Sheth, R. Fallahpour, R. Farran, Z. Halime, M. Sircoglou, A. Aukauloo, W. Leibl. Photochem Photobiol Sci 2022, 21, 247-259.
Photocatalysis of CO2 reduction
Faced with the urgent need to mitigate global climate change, scientists are challenged to reduce carbon dioxide (CO2) emissions by developing catalysts that can help capture, activate, and transform CO2 into energy-rich chemicals. To rationally design our new catalyst, we were inspired by active sites of carbon monoxide dehydrogenases (CODH). In these metalloenzymes, which reversibly reduce CO2 to CO with high efficiency and selectivity, the metallic active site has an optimized surrounding known as second coordination sphere composed of amino acid residues that induce weak interactions such as hydrogen bonds with CO2 to assist the metal center.
Learning from these chemical subtleties, we introduced various functional groups on the periphery of an iron porphyrin catalyst: urea as multipoint hydrogen bonding pillars, imidazolium as cationic moieties, and another nearby metalloporphyrin as bimetallic activator. Electrochemical, spectroscopic and crystallographic studies showed that the meticulous 3D positioning of these groups improves the energy efficiency of the catalytic CO2 reduction while maintaining high turnover frequency and selectivity. Further following inspiration from enzymatic processes, we valorized the reduction of CO2 to CO in carbonylation processes, opening interesting opportunities for the synthesis of industrial and pharmaceutical compounds.
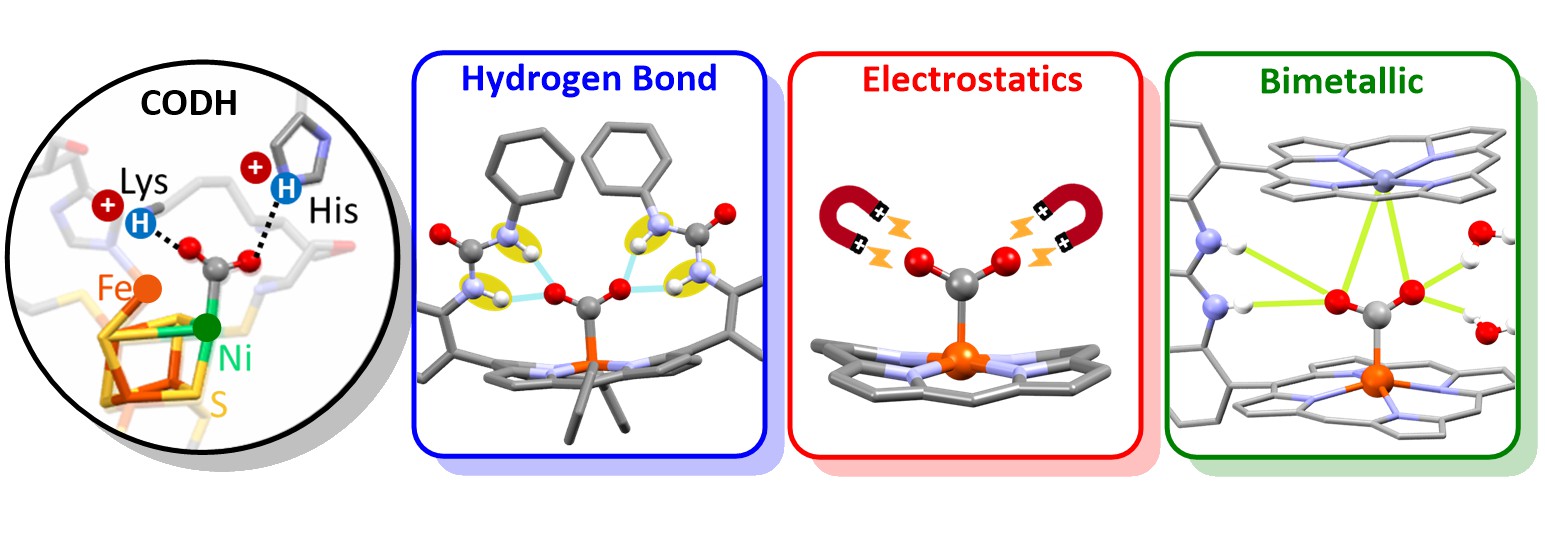
Representation of the active site of the CO dehydrogenase enzyme (left) and a modified iron porphyrin as bio-inspired model. © Gotico/CEA
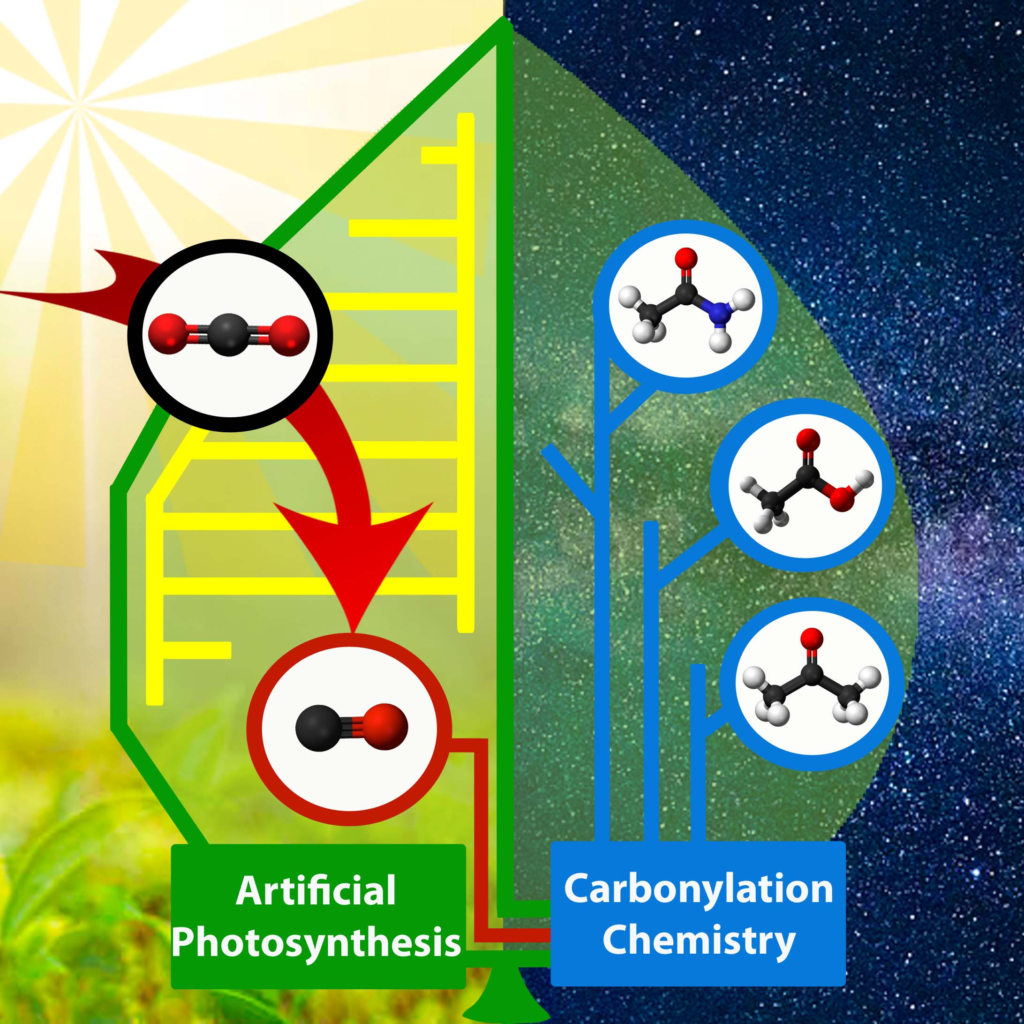
Representation of the valorization of CO produced from photocatalytic CO2 reduction to various carbonylation processes. © Gotico/CEA
Selected Publications
Bio-Inspired Bimetallic Cooperativity Through a Hydrogen Bonding Spacer in CO2 Reduction. C. Zhang, P. Gotico, R. Guillot, D. Dragoe, W. Leibl, Z. Halime, A. Aukauloo, Angewandte Chemie International Edition, 2023, 62, e202214665.
Dissection of Light-Induced Charge Accumulation at a Highly Active Iron Porphyrin: Insights in the Photocatalytic CO2 Reduction. E. Pugliese, P. Gotico, I. Wehrung, B. Boitrel, A. Quaranta, M.-H. Ha-Thi, T. Pino, M. Sircoglou, W. Leibl, Z. Halime, A. Aukauloo, Angewandte Chemie International Edition, 2022, 61, e202117530.
Atropisomeric Hydrogen Bonding Control for CO2 Binding and Enhancement of Electrocatalytic Reduction at Iron Porphyrins. P. Gotico, L. Roupnel, R. Guillot, M. Sircoglou, W. Leibl, Z. Halime, A. Aukauloo, Angewandte Chemie International Edition, 2020, 59, 22451-22455
Second-Sphere Biomimetic Multipoint Hydrogen-Bonding Patterns to Boost CO2 Reduction of Iron Porphyrins. P. Gotico, B. Boitrel, R. Guillot, M. Sircoglou, A. Quaranta, Z. Halime, W. Leibl, A. Aukauloo, Angewandte Chemie International Edition, 2019, 58, 4504-4509
Through-Space Electrostatic Interactions Surpass Classical Through-Bond Electronic Effects in Enhancing CO2 Reduction Performance of Iron Porphyrins. A. Khadhraoui, P. Gotico, W. Leibl, Z. Halime, A. Aukauloo, ChemSusChem 2021, 14, 1308–1315.
Local ionic liquid environment at a modified iron porphyrin catalyst enhances the electrocatalytic performance of CO2 to CO reduction in water. A. Khadhraoui, P. Gotico, B. Boitrel, W. Leibl, Z. Halime, A. Aukauloo, Chem. Commun., 2018, 54, 11630-11633
Unlocking full and fast conversion in photocatalytic carbon dioxide reduction for applications in radio-carbonylation. S. Monticelli, A. Talbot, P. Gotico, F. Caillé, O. Loreau, A. Del Vecchio, A. Malandain, A. Sallustrau, W. Leibl, A. Aukauloo, F. Taran, Z. Halime, D. Audisio, Nature Communications, 2023, 14, 4451
Visible Light-Driven Reduction of CO2 to CO and Its Subsequent Valorization in Carbonylation Chemistry and 13C Isotope Labelling. P. Gotico, A. Del Vecchio, D. Audisio, A. Quaranta, Z. Halime, W. Leibl, A. Aukauloo, ChemPhotoChem, 2018, 2, 715-719
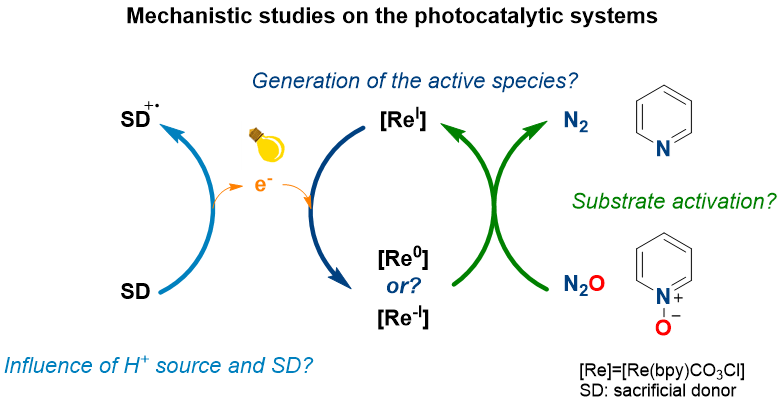
Photocatalysis of NO and SO bond deoxygenation
The deoxygenation of N–O and S–O bonds is a chemical challenge, and is key to provide interconversion pathways between various oxidation states of N and S containing compounds. Such processes are necessary to close the nitrogen and sulfur cycles which are at the moment open due to human activities. On a synthetic point of view, the development of selective methods for the deoxygenation of N–O and S–O bonds will allow shifting the source of nitrogen and sulfur in industrial processes. These reactions are currently relying on thermochemical methods and sacrificial reducing agents. We are interested in studying new catalytic methods for the deoxygenation of N–O and S–O bonds involving selective and efficient photo- and electro transition metal based-catalysts. Mechanistic investigation using time-resolved visible and FTIR spectroscopy are used for mechanism elucidation for a rational design of efficient methods.
Photocatalysis of water oxidation
In artificial photosynthesis, chemists are aiming to borrow principles from natural photosynthesis to develop photoelectrochemical cells for water splitting. The water plastoquinone photooxidoreductase enzyme, also known as Photosystem II, uses light to perform the four-electron, four-proton oxidation of water to dioxygen and stores reducing equivalents in reduced forms of quinones which are ultimately used in dark reactions for the synthesis of energy-rich molecules.
Despite considerable efforts, functioning molecular models of photosystem II are still elusive. Among different materials under intense investigation for this thrust, organic semiconductors have captured increasing attention for their synthetic readiness, robustness and handles to adjust the energy levels of the conduction and valence bands. We have recently reported that visible light irradiation of a suspension of nano-structured conjugated polymers based on PolyDiPhenylButadiyne (nano-PDPB herein) in water leads to O2 evolution. The electrons (and protons) extracted from water are stored on the nanostructured semiconductor and can be recovered as reduced form of quinone paralleling the functions of Photosystem II (Scheme 1).

Selected Publications
Visible light-driven simultaneous water oxidation and quinone reduction by a nano-structured conjugated polymer without co-catalysts. J. Patel, X. Yuan, S. M. Marinho, W. Leibl, H. Remita, A. Aukauloo, Chem. Sci., 2020
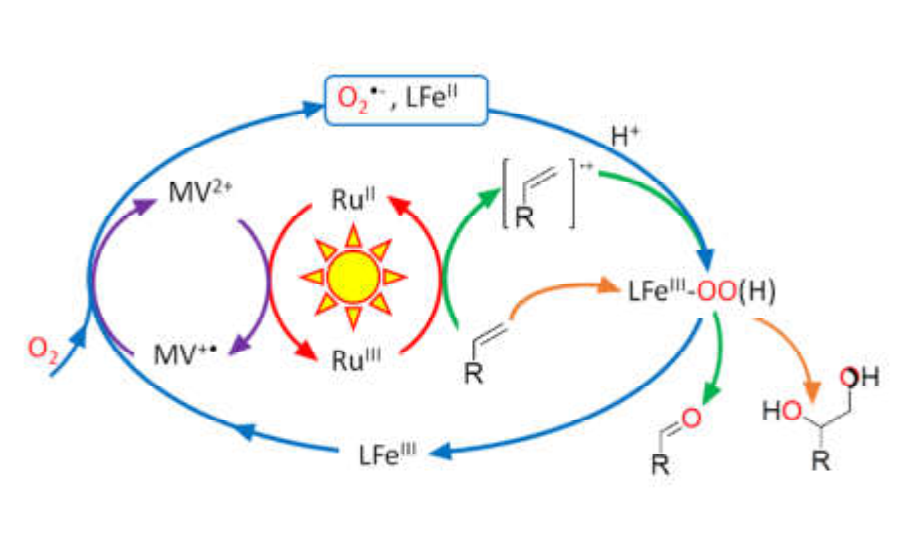
Reductive activation of O2
Bio-inspired approaches to artificial photosynthesis play a very important role in the sustainable development of the chemical industry. In nature, oxygen from photosynthesis is used, among other things, to oxidize organic substrates essential to biology and chemistry. In this context chemists aim to reproduce this optimal method: they intend to use solar energy and oxygen to activate catalysts that will then be able to realize the oxygen atom transfer reactions necessary for the manufacture of organic substrates useful in all industrial sectors.
However, current methods require the addition of molecules acting as “sacrificial electron donors”, which limits the efficiency of the photocatalytic reaction. Indeed, it is a chemical species that provides electrons but whose presence can interfere in the desired reaction and which leads to undefined by-products.
We showed that the use of a reversible electron acceptor, methylviologen (MV), widely used in photo-induced electron transfer studies, overcomes this limitation. MV serves as a relay in the electron transfer to O2 and the catalyst (iron complex) generating the reactive species for the desired catalysis (Scheme).
One more step towards efficient and clean photocatalysis
Selected Publications
A Reversible Electron Relay to Exclude Sacrificial Electron Donors in the Photocatalytic Oxygen Atom Transfer Reaction with O2 in Water. N. T. Vo, Y. Mekmouche, T. Tron, R. Guillot, F. Banse, Z. Halime, M. Sircoglou, W. Leibl, A. Aukauloo, Angewandte Chemie International Edition, 2019, 58, 16023-16027.
Photocatalytic generation of a non-heme Fe(III)-hydroperoxo species with O2 in water for the oxygen atom transfer reaction. E. Pugliese, N.T. Vo, A. Boussac, C. Herrero, F. Banse, Y. Mekmouche, J. Simaan, T. Tron, P. Gotico, M. Sircoglou, Z. Halime, W. Leibl, A. Aukauloo. Chem. Sci. 2022, 13, 12332-12339,