In numerous eukaryotes, developmentally programmed elimination of germline DNA drives the plasticity of the somatic genome. Because of its unique nuclear dimorphism, Paramecium provides an extraordinary unicellular model to study the impact of transposable elements (TEs) on genome dynamics.
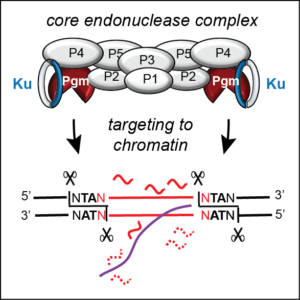
A major research line of our team aims at understanding the molecular mechanisms involved in programmed DNA elimination and its epigenetic control. We identified several key actors of IES excision. The PiggyMac endonuclease, a catalytically active domesticated PiggyBac transposase, introduces programmed DNA double-strand breaks (DSB) at IES ends and initiates their precise excision. PiggyMac works in association with five different domesticated transposases from the same family. We use molecular, biochemical and cellular approaches combined with reverse genetics and high-throughput next-generation sequencing to characterize the endonuclease-associated complex, the function of its different subunits and the way it recognizes its chromatin targets.
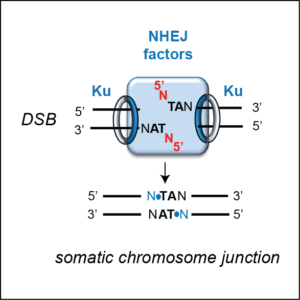
Precise non-homologous end joining
DSBs are potentially very deleterious DNA lesions that threaten genome stability and, if not repaired correctly, may cause cancer or cell death. They are repaired through two cellular pathways: homologous recombination or non-homologous end joining (NHEJ). In Paramecium, DSB introduction by the PiggyMac-associated complex is essential for the assembly of a functional somatic genome. The classical NHEJ pathway, involving a development-specific Ku70/Ku80 heterodimer and the Ligase IV/Xrcc4 complex, mediates the precise repair of IES excision sites and allows functional genes to be assembled in the new MAC. We showed that DNA cleavage at IES ends strictly depends upon the presence of Ku. One of our goals is to study how the endonuclease complex interacts with NHEJ factors to ensure that programmed rearrangements do not jeopardize genome integrity.
Paramecium genomics
The model species Paramecium tetraurelia belongs to the aurelia complex of 15 sibling species. These species diverged after a whole genome duplication and provide a powerful system to test hypotheses about speciation/genetic (in)compatibility. Our team’s bioinformaticians have been involved in the sequencing and annotation of germline genomes of P. aurelia species and outgroups, in order to elucidate Paramecium chromosome organization and study the evolutionary dynamics of IESs and repeated sequences (transposable elements, minisatellites).
Ongoing functional genomics projects involve the development of dedicated bioinformatics pipelines to analyze high-throughput DNA and RNA sequencing data. Major objectives are to (i) monitor the time-course of DNA elimination during MAC development at the genome-wide scale; (ii) characterize the epigenetic determinants of the recognition of eliminated DNA and (iii) study the impact of genome rearrangements on the transcriptional program.
Paramecium Database
The team developed and curates the model organism database ParameciumDB, a unique information resource essential to the Paramecium research community and a service of ELIXIR France since 2019. ParameciumDB is a user-friendly information system with a public web interface that provides high-quality, curated resources including annotations for Paramecium genomes, integrated with functional datasets (transcriptomic, proteomic, gene silencing) and bibliography. Advanced query and data retrieval tools and interactive genome browsers are available. Current efforts aim to extend comparative genomics tools as more genomes are sequenced and incorporated.