Senescence and Genome Stability
Cellular senescence is a stress response of mammalian cells leading to a stable proliferative arrest associated with a characteristic transcriptome including the expression of inflammatory genes. We study cellular senescence as a tumor suppressor mechanism that may also contribute to aging.
Cellular senescence as a tumor suppressor mechanism
Senescence can be triggered in conditions associated with genomic instability such as the loss of telomeric sequences when somatic human cells undergo excessive proliferation with insufficient telomerase expression. Senescence can also be triggered by the expression of hyper-mitogenic oncogenes. The development of a tumor requires the expression of oncogenes that provoke uncontrolled proliferation. Remarkably however, the expression of oncogenes such as RASval12 or B-RAF-V600E can trigger senescence when expressed in normal human cells. Thus, for a tumor to develop, senescence effector pathways involving the tumor suppressors p53 and Rb must be inactivated. How can oncogene expression trigger senescence? The production of reactive oxygen species and DNA damage associated with replicative stress have been implicated in senescence induced by RASval12 expression. However, we found that RAF-induced senescence can occur without apparent oxidative stress, DNA damage or DNA replication (Jeanblanc et al. Oncogene. 2012). We are thus interested in defining novel pathways contributing to RAF-induced senescence. The B-RAF-V600E oncogenic mutation is a major driver mutation in melanoma and also contributes significantly to several other human cancers including thyroid and colorectal carcinomas. We have produced a model human fibroblast cell line in which we can modulate the expression of B-RAF-V600E to study the pathways leading to RAF-induced senescence. Hypothesis-based and systems biology approaches are being used to identify pathways. The importance of these pathways will be confirmed using a mouse model in which conditional expression of B-RAF-V600E leads to melanocyte senescence.
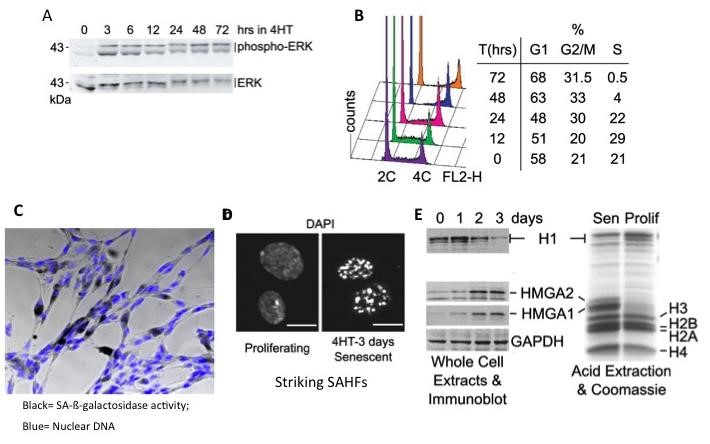
Figure 1: conditional hyperactivation of the RAF kinase in human fibroblasts leads to a rapid hyperactivation of the downstream ERK1/2 MAP kinases (A) followed by a strong inhibition of DNA synthesis by 2 days (B) and the appearance of characteristic markers of cellular senescence, including senescence-associated ß-galactosidase activity (C), senescent-associated heterochromatic foci (D), and the accumulation of the non-histone chromatin proteins HMGA1 and HMGA2 with a decrease of histone H1 isoforms (E).
The histone variant H2A.J accumulates in senescent cells
and promotes inflammatory gene expression
The senescence of mammalian cells is characterized by a proliferative arrest in response to stress and the expression of an inflammatory phenotype. Chromatin modifications have been implicated in the stable repression of proliferative genes in senescence, and in the derepression of inflammatory genes. We used mass spectrometry to characterize the chromatin of senescent cells (Contrepois et al. J. Proteome Res., 2010 and Contrepois et al. Epigenetics & Chromatin, 2012). We discovered that H2A.J, a poorly studied H2A variant found only in mammals, accumulates in human fibroblasts in senescence with persistent DNA damage. H2A.J also accumulates in mice with aging in a tissue-specific manner. Knock-down of H2A.J inhibited the derepression of inflammatory genes that contribute to the senescent-associated secretory phenotype (SASP), and over-expression of H2A.J increased the expression of some of these genes in proliferating cells. H2A.J accumulation may thus promote the signaling of senescent cells to the immune system, and it may contribute to chronic inflammation and the development of diseases with aging. We are currently investigating the mechanism by which H2A.J promotes inflammatory gene expression in senescent cells, and we are interested in studying its functions in tissues in which H2A.J accumulation does not appear to be linked to senescence.
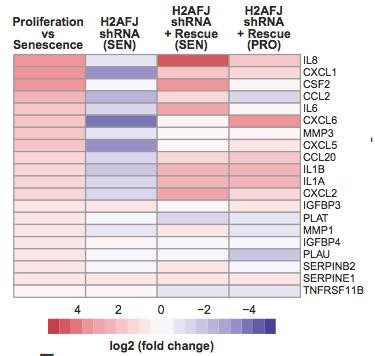
Figure 2: heat map from transcriptome analyses showing that knock-down of H2AFJ inhibits the derepression of inflammatory genes in senescent human fibroblasts.
Small molecule/natural product modulators of cellular senescence
The accumulation of senescent cells has been implicated in various human pathologies associated with stress or aging. Senescence may interfere with tissue regeneration by limiting the proliferation of stem cells, and the secretion of inflammatory factors by senescent cells may have negative paracrine effects associated with chronic inflammation. Mouse models have indicated that the genetic elimination of senescent cells may limit some phenotypes associated with aging. We are screening for small molecules/natural products that modulate the senescent phenotype in collaboration with the Chemistry department and the PARi screening platform at the CEA/Saclay. Molecules that bypass or revert senescence, that inhibit the inflammatory phenotype, or that are specifically toxic for senescent cells are all of interest (figure 3). These molecules may serve as probes to dissect senescence mechanisms, and they may represent lead compounds for modulating senescence in human diseases. A pilot screen of the Prestwick chemical library composed of FDA-approved drugs revealed a profound effect of a specific class of compounds on RAF-induced senescence. We are currently performing transcriptome analyses to determine how these molecules modulate senescence.
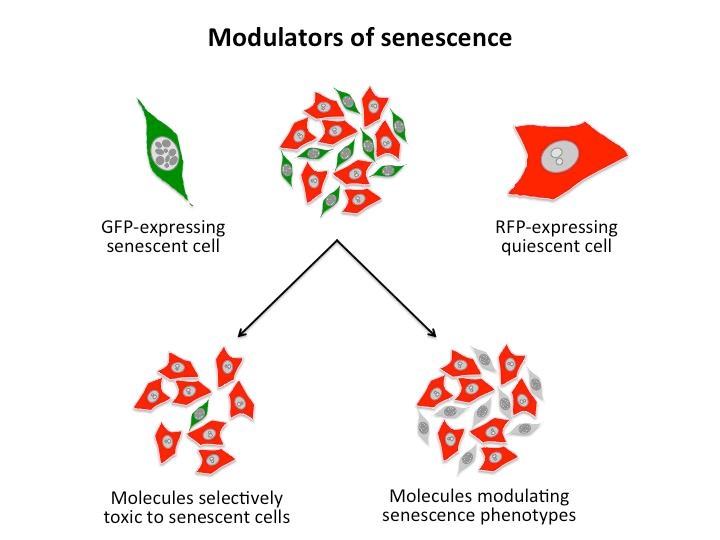
Figure 3: Possible screening concepts for modulators of senescence
team

Group Leader Researcher

Technician
Latest publications
For all the publications of the Team click on the button below.
External funding
![]()