Clostridium difficile CRISPR-Cas system function and regulation
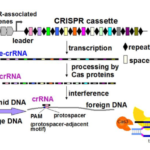
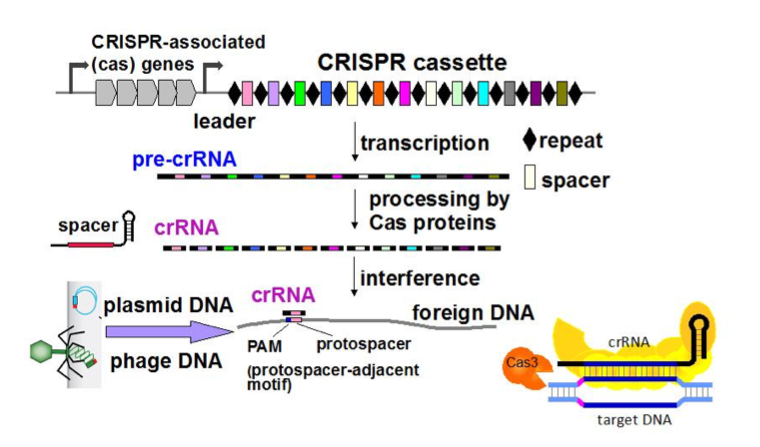
During infection, Clostridium difficile survives in phage-rich gut communities by relying on efficient anti-invader defence systems including CRISPR-Cas for prokaryotic adaptive immunity. CRISPR arrays are composed of repeat sequences separated by spacers matching with and originating from foreign DNA. They are transcribed into a pre-crRNA, which is cleaved by the Cas proteins to give CRISPR RNAs (crRNAs). In complex with Cas proteins, crRNAs interfere with phage infection and plasmid conjugation by recognizing foreign DNA and targeting it for destruction. crRNAs are among the most abundant ncRNAs detected by RNAseq (Soutourina et al, PLos Genetics, 2013). The originality of C. difficile CRISPR-Cas system is the presence of an unusually large set of CRISPR arrays, the presence of 2 sets of cas genes and the prophage location of several CRISPR arrays. We have recently shown active expression and processing of CRISPR RNAs from multiple CRISPR arrays and provided experimental evidence for CRISPR-Cas system functionality in C. difficile. Through genome sequencing and host-range analysis of several new C. difficile phages and plasmid conjugation experiments we demonstrated the defensive function of CRISPR-Cas system in both reference and epidemic C. difficile strains. Altogether, these data emphasize the original features of active C. difficile CRISPR-Cas system that might be important for C. difficile survival during its infection cycle (Boudry et al, mBio, 2015). This sets the stage for further mechanistic and physiological analyses of CRISPR-Cas-mediated interactions of important human pathogen with its genetic parasites.
We have recently shown the possibility of redirecting this endogenous CRISPR-Cas system to autoimmunity, which allows efficient editing of the C. difficile genome. The new method expands the arsenal of genetic tools available for the study of C. difficile and can serve as a basis for new strategies to fight C. difficile infections (Maikova et al, AEM, 2019; Maikova et al, Frontiers in Microbiology, 2018).
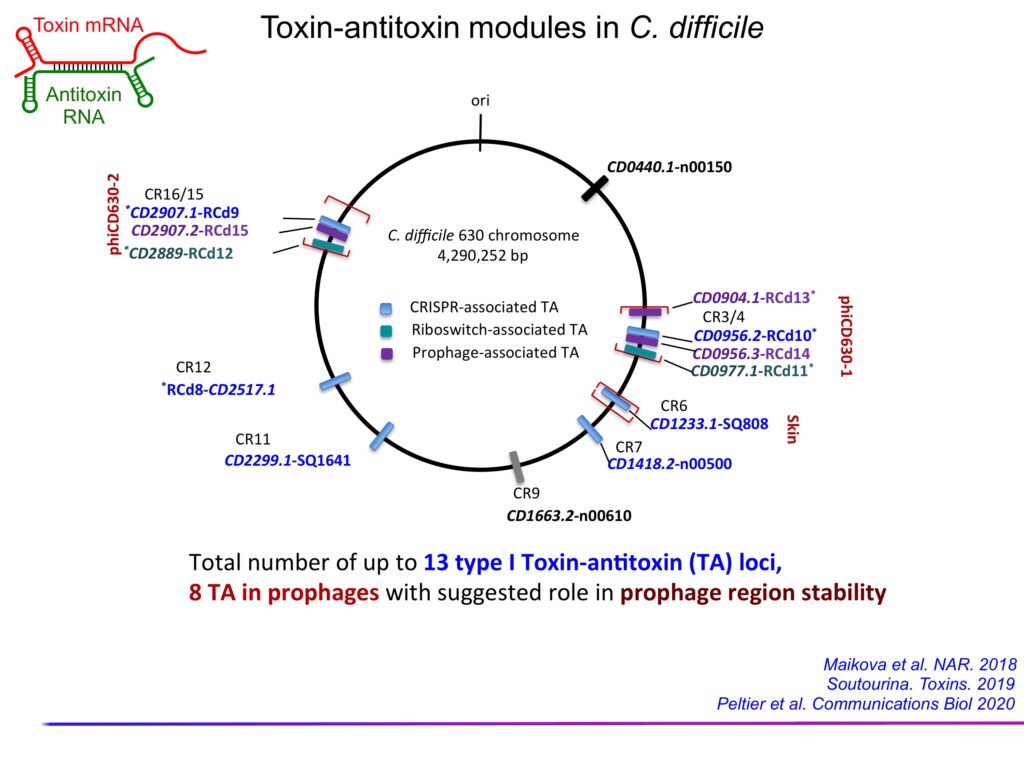
Toxin-antitoxin (TA) modules are widespread in bacteria and archaea. These loci include two genes encoding a stable toxin and an unstable antitoxin. Overexpression of the toxin has bactericidal or bacteriostatic effects on the host cell, while the antitoxin is capable of neutralizing the action or production of the toxin. In Type I systems, the antitoxin is an antisense RNA that targets the toxin’s mRNA for degradation and/or inhibition of translation. We have identified the first type I TA systems associated with CRISPR cassettes in C. difficile genomes (Maikova et al, NAR, 2018). The co-localization and co-regulation of the TA and CRISPR systems suggest a possible genomic link between these systems of cell dormancy and adaptive immunity. A total of 13 type I TA modules are present in the genome of the reference strain 630, several of which located in C. difficile prophages contribute to the maintenance and stability of mobile elements (Soutourina, Toxins, 2019; Peltier et al, Communications Biology, 2020). The antitoxins in these systems represent the first functional antisense RNAs identified in C. difficile. We have used inducible toxicity caused by a type I toxin as a counter-selection marker in the genome editing procedure by allelic exchange. This promotes the elimination of plasmid-bearing cells, which greatly improves genome editing efficiency (Peltier et al, Communications Biology, 2020).
CloSTARn : Role of the c-di-GMP signaling pathway
during the Clostridium difficile infection cycle
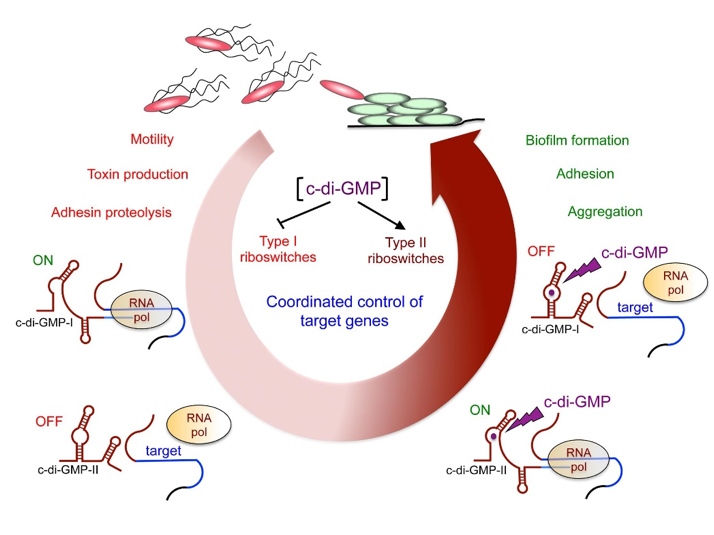
Among the ncRNA identified, we are particularly interested in the riboswitches responding to an important signaling molecule: cyclic-di-guanosyl-5’monophosphate (c-di-GMP). C-di-GMP controls lifestyle switch from free-living motile state to biofilm communities and virulence in bacteria. In contrast to most Gram-positive bacteria, C. difficile encodes a large number of c-di-GMP turnover enzymes (up to 37) suggesting an important role for this molecule. The originality of c-di-GMP-dependent control in C. difficile is the use of RNA molecules as effectors instead of protein effectors found in the majority of bacterial systems. We have detected the expression of 16 predicted c-di-GMP-dependent riboswitches that negatively control downstream gene expression by premature termination of transcription or positively modulate target gene translation and demonstrated their functionality (Soutourina et al, PLoS Genetics, 2013). We will further investigate their role in the coordinated control of processes crucial for successful development of C. difficile inside the host such as motility, adhesion, biofilm formation, and resistance to phage infection.
Involvement of Hfq in the ncRNA regulatory network
In many organisms, trans-acting ncRNAs require the RNA chaperone protein Hfq for their action. Hfq can stabilize ncRNAs and their interactions with target mRNAs leading to negative or positive modulation of gene expression or can induce ncRNA cleavage promoting degradation of associated mRNAs. We recently demonstrated that Hfq depletion had pleiotropic effects in C. difficile, an unique feature in Gram-positive bacteria (Boudry et al, J. Bacteriol., 2014). In C. difficile, Hfq is involved in the control of metabolic adaptations, biofilm formation, stress responses and sporulation. The accumulation of several ncRNAs was altered when Hfq is depleted and Hfq can bind selected ncRNAs supporting its involvement in their function. We are currently analyzing the Hfq-dependent RNAs controlling several key steps of C. difficile infection cycle such as sporulation process, stress response and biofilm formation. Several ncRNAs are specific of epidemic strain 027 or differentially expressed compared to reference strain 630. By combining in silico, in vitro and in vivo approaches, we will study the function of the most interesting candidate ncRNAs to see their possible involvement in the hypervirulence of this worldwide epidemic strain. We will further explore the role of the 027-specific ncRNAs in pathogenesis-related processes by analyzing the effects of their inactivation/overexpression on various phenotypes and potential targets. Then we will study the in vitro interactions of these ncRNAs with their mRNA targets, the role of Hfq in the regulatory processes and further their role during infection in C. difficile animal models for both colonization and virulence.
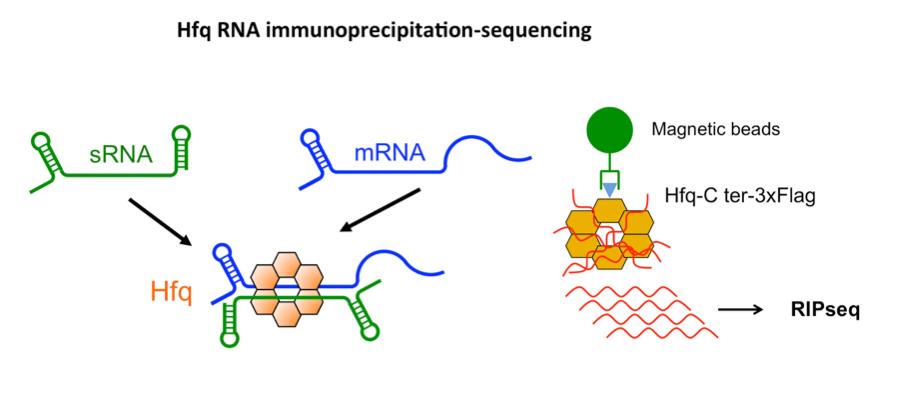
We performed a high-throughput RNA sequencing analysis by immunoprecipitation (RIP-Seq) to identify Hfq-associated RNAs in C. difficile. Our work revealed a large set of ncRNAs and mRNAs interacting with Hfq, including leader mRNAs and coding regions, known regulatory RNAs and potential new ncRNAs (Boudry et al, RNA Biology, 2021). These data provide an essential molecular basis for further studies on the post-transcriptional regulatory network of this emerging enteropathogen.