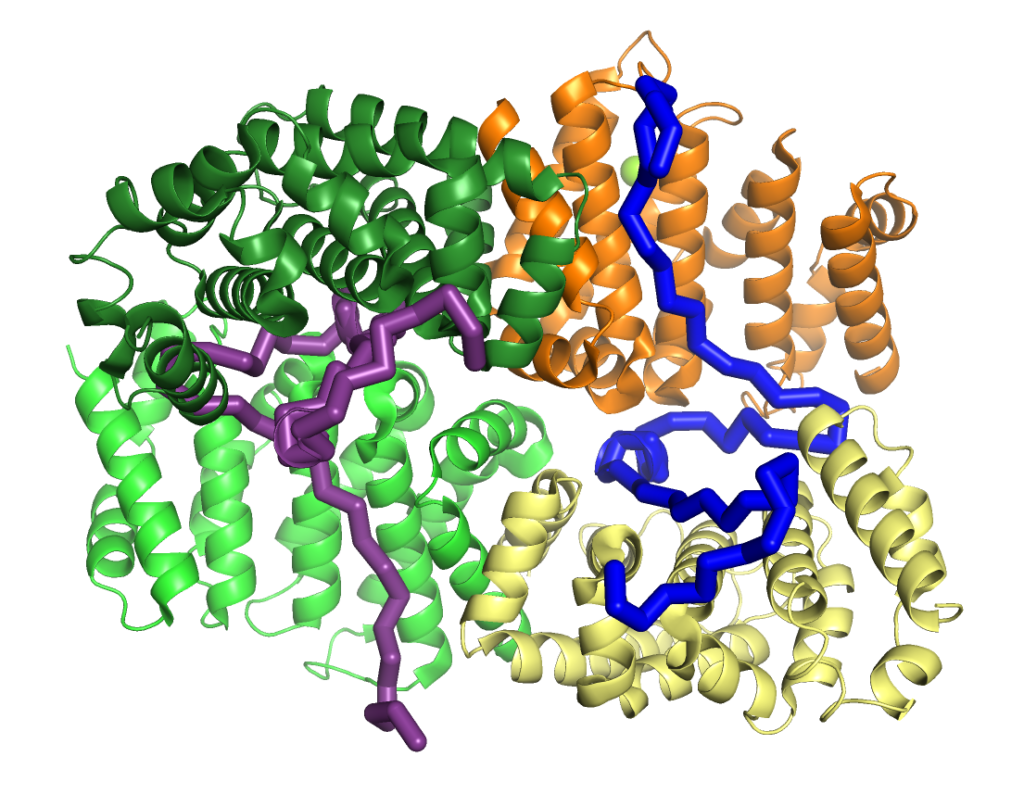
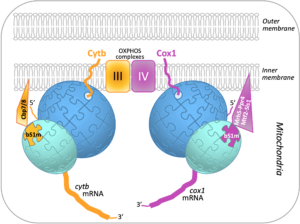
Mitochondria contain their own DNA and translation machinery. Mitochondrial mRNAs encode key subunits of the oxidative phosphorylation (OXPHOS) complexes, that produce energy for the whole cell. Thus, mitochondrial translation defects lead to severe diseases in humans. The fission yeast Schizosaccharomyces pombe is a valuable model to study mitochondrial gene expression, since it closely resembles humans in its mitochondrial DNA structure and physiology. By combining bioinformatics, genetic and biochemical approaches including mass spectrometry on the I2BC SICaPS platform, the I2BC BIOMIT group discovered two interacting factors of S. pombe, Cbp7 and Cbp8, controlling the production of Cytb, a catalytic subunit of OXPHOS complex III. Two classes of Cbp7/Cbp8 partners were identified and shown to modulate the synthesis of Cytb or Cox1, key subunits of the OXPHOS complexes III and IV respectively. First, two isoforms of bS1m, a protein of the small mitoribosomal subunit, that appear mutually exclusive and confer translational specificity. Second, a complex of four proteins dedicated to Cox1 synthesis, which includes an RNA helicase, Mrh5, that interacts with the mitochondrial ribosome. These data suggest that S. pombe contains, in addition to complexes of translational activators, a heterogeneous population of mitochondrial ribosomes that could specifically modulate translation depending on the mRNA translated, in order to optimally balance the production of different respiratory complex subunits. Together, these results, published in Nucleic Acids Research, support the view that ribosomes are not merely translation machines, but can also behave as regulatory elements.
Contact: Nathalie Bonnefoy – Team SICaPS (nathalie.bonnefoy@i2bc.paris-saclay.fr)

After completing a PhD at Indiana University in the 1970s and a post-doc at Cold Spring Harbor under the supervision of Jim Watson, Michael Dubow started his career in 1980 as Assistant Professor at Mac Gill University in Canada, then as Associate Professor in 1987 and full Professor in 1991 in the Department of Microbiology and Immunology and the Department of Human Genetics at this University. After having obtained a 2-year availability at the University of Metz, between 1996 and 1998, he settled permanently in France in 2002, creating a research team at the Institute of Genetics and Microbiology and then in 2015 at the Institute for Integrative Biology of the Cell (I2BC).
The work carried out and developed under his direction by his team at the IGM has initiated innovative research in the field of microbial ecology. Indeed, important data concerning the microbial and phage diversity in oligotrophic environments, little studied until now, have been obtained. Thanks to this work, an unsuspected diversity in extreme environments, such as deserts, has been revealed. Working on phage particles directly from desert sand samples represented a real technical challenge to extract them without disrupting their structures. Functional metagenomics from the same type of samples also allowed the isolation of new enzymes.
Throughout his career, Michael Dubow was always forward-thinking, constantly showing his interest in genomics, phage therapy, phage and bacterial biodiversity and his interest in applications (such as water quality ….). He was a very open-minded teacher-researcher, following the evolution of many disciplinary fields. He worked hard and “militated” for the development of interdisciplinary research “at the interfaces”, in particular by participating in the creation of the master’s degree “Physics of biological systems” or by proposing an optional course of the same name in the second year of the biology degree. He also taught courses in “Life Science for Engineers” at Supélec and CentraleSupélec. His interest in the physics-biology interface is currently being pursued with the development of a subject on the study of the degradation mechanisms of biofilms by cold plasma at atmospheric pressure (in collaboration with the GeePs team at CentraleSupélec).
He was the author of more than 110 publications during his career. He was a Fellow of the American Academy of Microbiology since 1994 and of the Royal Society of Medicine (UK) since this year. He was also the author of several patents and participated in the creation of 3 companies (Bomec Inc.; PhageTech/Targanta Therapeutics Inc.; Biolumine SA).
Michael was also recognized as an outstanding teacher, appreciated by the students for his lively lectures (expert in mime to illustrate the molecular biology of phages), always ready to welcome foreign PhD students in his team, actively participating in the delocalized Master in Vietnam at the University of Ho Chi Minh City and in the Master 2 defenses of the Vietnamese students by videoconference, but also to the students at the Physics-Biology Interface. Michael had a subtle way of arousing the curiosity of his students, he knew how to tease them and push them to think by provoking them, but always with kindness. He often challenged them by saying during a lecture “you believe or you think?…belief is for Sunday, thought is for science”.
Michael was also recognized for his investment in the University of Paris-Sud, as an elected member and then as Vice-president for research of the Biology Department, but also in the scientific council of the Faculty of Sciences, in the international relations service of the University where he regularly carried out missions in South-East Asia. He also held national missions as a member of the CNU65 and the Scientific Council of the Institute of Biological Sciences (INSB) of the CNRS.
Beyond his qualities as a researcher and teacher, Michael Dubow was a deeply kind man. He always had a smile on his face, was always willing to help, and was a careful proofreader of many papers for several teams, correcting our mistakes in English. Whatever the bad news, he was resolutely positive, supporting his colleagues in joy and sorrow, often warning us against excessive Americanization of our society, as he knew it so well. For those who had the chance to meet him on a daily basis, “Mike” was always delighted to chat about science of course, but also about all the subjects of life such as sports (Ha! the American Baseball Championship…), music (especially Jazz), movies (he was a real Star Wars fan), politics.
Like all humans, Mike had a complex nature and it is difficult to sum up this complexity in a few words, but he was a lover of life…one of his favorite phrases that best characterized him was “la vie est belle “.
His sudden death will leave a great void at the University of Paris-Saclay. His tall figure, his accent, his prolific scientific ideas will be terribly missed, but he was able to plant enough seeds here and there, so that his ideas and his spirit will live on.
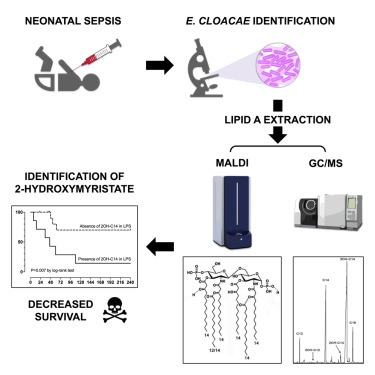
 The nitrogen fixing symbiosis of legumes with rhizobium bacteria has a predominant ecological role in the nitrogen cycle and has the potential to provide the nitrogen required for plant growth in agriculture. The host plants allow the nitrogen-fixing rhizobia to colonize the cells of specific symbiotic organs, the nodules, in very large numbers in order to produce sufficient reduced nitrogen for the plant needs. Some legumes, including Medicago spp., produce massively antimicrobial peptides to keep this large bacterial population in check. These peptides, known as NCRs, have the potential to kill the rhizobia but in the nodule cells, they rather inhibit the division of the endosymbionts and trigger them into a morphologically differentiated state, resulting in a high nitrogen fixing activity. In this study published in mBio, the Plant-Bacteria Interactions team of I2BC shows that the bacterial resistance to the antimicrobial activity of the NCR peptides in the Medicago symbiont Sinorhizobium meliloti is multifactorial and requires peptide transporters, the lipopolysaccharide outer membrane and the stress response regulator RpoH1.
The nitrogen fixing symbiosis of legumes with rhizobium bacteria has a predominant ecological role in the nitrogen cycle and has the potential to provide the nitrogen required for plant growth in agriculture. The host plants allow the nitrogen-fixing rhizobia to colonize the cells of specific symbiotic organs, the nodules, in very large numbers in order to produce sufficient reduced nitrogen for the plant needs. Some legumes, including Medicago spp., produce massively antimicrobial peptides to keep this large bacterial population in check. These peptides, known as NCRs, have the potential to kill the rhizobia but in the nodule cells, they rather inhibit the division of the endosymbionts and trigger them into a morphologically differentiated state, resulting in a high nitrogen fixing activity. In this study published in mBio, the Plant-Bacteria Interactions team of I2BC shows that the bacterial resistance to the antimicrobial activity of the NCR peptides in the Medicago symbiont Sinorhizobium meliloti is multifactorial and requires peptide transporters, the lipopolysaccharide outer membrane and the stress response regulator RpoH1.
https://doi.org/10.1128/mBio.00895-21
Contact: Peter MERGAERT (peter.mergaert@i2bc.paris.saclay.fr)

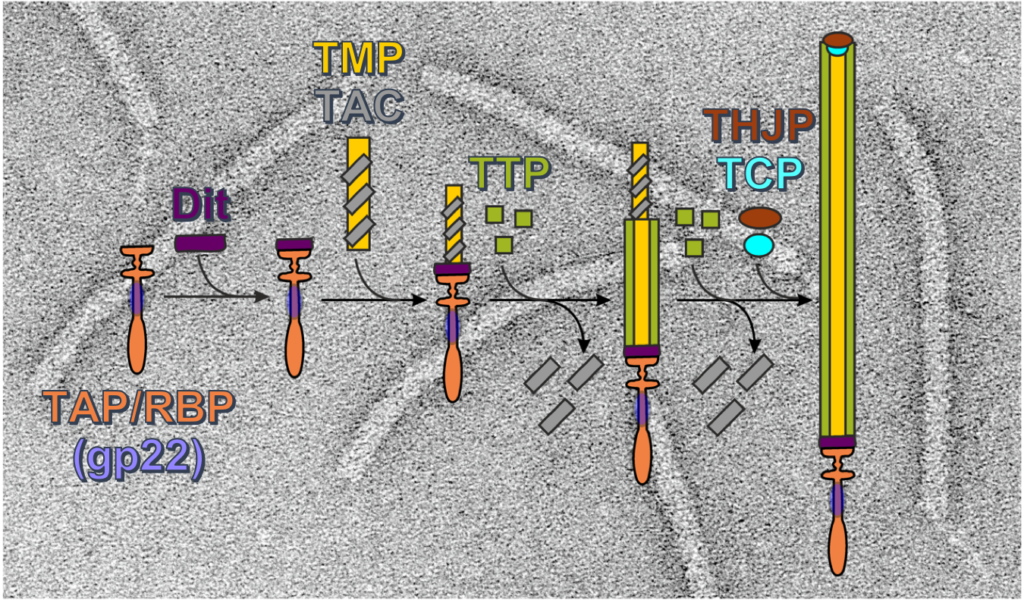
 Virus lytic infection imposes a major biosynthetic effort to the host cell and takes over significant cellular space. Viruses of prokaryotes must meet the challenge to restructure the cytoplasm open space of a small-sized cell. A study published in the Proceedings of the National Academy of Sciences USA reports the discovery that bacteriophage SPP1 infection leads to biogenesis of two types of membraneless compartments in the cytoplasm of the bacterium Bacillus subtilis. One is a single viral DNA compartment and others are warehouses for storage of viral particles. These compartments are temporal and spatially independent.
Virus lytic infection imposes a major biosynthetic effort to the host cell and takes over significant cellular space. Viruses of prokaryotes must meet the challenge to restructure the cytoplasm open space of a small-sized cell. A study published in the Proceedings of the National Academy of Sciences USA reports the discovery that bacteriophage SPP1 infection leads to biogenesis of two types of membraneless compartments in the cytoplasm of the bacterium Bacillus subtilis. One is a single viral DNA compartment and others are warehouses for storage of viral particles. These compartments are temporal and spatially independent.
The DNA compartment sequesters machines operating viral DNA transactions. Multiple hybrid DNA replication centers, containing both phage replication proteins and hijacked bacterial replisomes, operate at different sub-locations within the compartment for parallelized synthesis of viral DNA. DNA is subsequently packaged in virion precursors without DNA (procapsids) at the compartment edges. Viral DNA-filled capsids then segregate from the DNA compartment, bind phage tails, and the resulting virions build warehouse compartments.
This spatial partition of the B subtilis cell responds to the requirements for exponential replication of SPP1 genomes and for the assembly of hundreds of viral particles. Its similarities to remodelling of the cell nucleus by herpesviruses led to the hypothesis that ancestral strategies used by viruses to invade the cell space were conserved to infect hosts of different Domains of Life.
Part of this work is from the PhD thesis of Audrey Labarde at Université Paris-Saclay.
https://doi.org/10.1073/pnas.2018297118
Contact: Paulo TAVARES (paulo.tavares@i2bc.paris.saclay.fr)

 Photosynthesis begins with the absorption of light energy by the chlorophyll pigments of the light collecting antennae (LHC – Light Harvesting Complex, complexes composed of proteins and chlorophyll and carotenoid pigments). This absorption creates an excitation energy (passage from a fundamental electronic state to an excited state of the collecting chlorophyll), energy which is transferred from one chlorophyll to another, to the reaction center of photosynthesis where it is converted into chemical potential energy (by charge separation). This energy conversion process is extremely efficient. So efficient, that it can cause a potentially deleterious overexcitation of the system. The plant sets up mechanisms to protect itself from this: from the macroscopic scale, by the movement of its leaves, to the molecular scale, by a mechanism that allows the dissipation of energy in the form of heat. This last mechanism, multifactorial, is called non photochemical quenching of chlorophyll fluorescence.
Photosynthesis begins with the absorption of light energy by the chlorophyll pigments of the light collecting antennae (LHC – Light Harvesting Complex, complexes composed of proteins and chlorophyll and carotenoid pigments). This absorption creates an excitation energy (passage from a fundamental electronic state to an excited state of the collecting chlorophyll), energy which is transferred from one chlorophyll to another, to the reaction center of photosynthesis where it is converted into chemical potential energy (by charge separation). This energy conversion process is extremely efficient. So efficient, that it can cause a potentially deleterious overexcitation of the system. The plant sets up mechanisms to protect itself from this: from the macroscopic scale, by the movement of its leaves, to the molecular scale, by a mechanism that allows the dissipation of energy in the form of heat. This last mechanism, multifactorial, is called non photochemical quenching of chlorophyll fluorescence.
Whether in vivo or in vitro, the Membrane Bioenergetics and Stress team (I2BC department), led by Bruno Robert, has shown that this quenching is linked to a rearrangement of the proteins and pigments that make up the LHC that creates “energy traps”. The excited chlorophyll pigments transfer their energy to carotenoid pigments which immediately dissipate it as heat. In the case of LHCII, the main collecting antenna of higher plants, in vitro spectroscopy experiments conducted on the aggregated complex (in the absence of the detergent conventionally used to solubilize it) establish that this transfer occurs between chlorophyll a and a lutein (a carotenoid). The extent of the extinction seems to be correlated with conformational changes (torsion) affecting lutein and another carotenoid, neoxanthin.
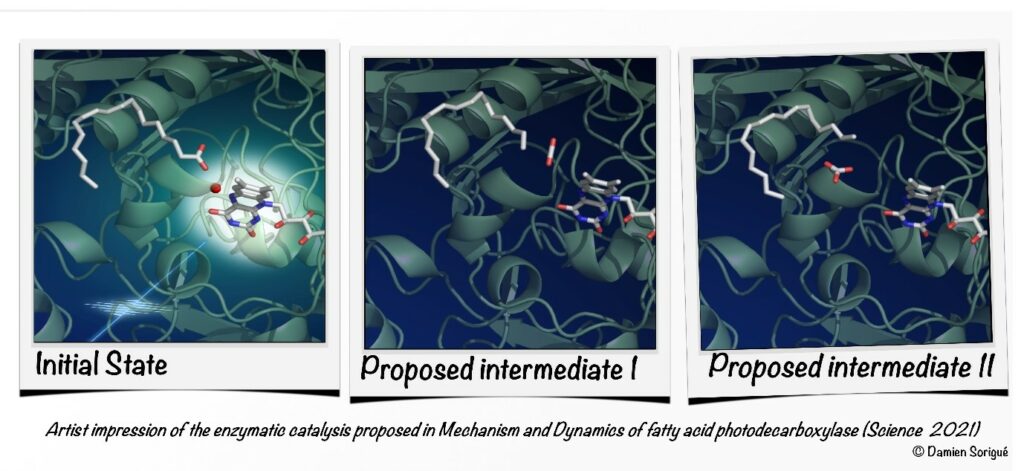
To further describe and understand these changes, Bruno Robert’s team studied the structure of LHCII in different environments that influence its electronic properties. Remarkably, the team succeeded in isolating for the first time a state of LHCII, obtained using the detergent n-dodecyl-α-D-maltoside, and in characterizing the spectroscopic properties of its pigments. Their study, published in JBC, shows that in this state all the changes associated with non-photochemical quenching (changes in protein-chlorophyll interactions, neoxanthin twist) are present except for lutein twist, while no quenching is associated with this state. This state of LHCII would be in some way an intermediate state that would allow the transition from a “lit” state, capable of absorbing and converting light energy into chemical energy, to a “quenched” state by non-photochemical quenching. The neoxanthin twist would be an indicator of large-scale conformational changes in LHCII that would precede smaller-scale changes directly responsible for quenching and revealed by the lutein twist. This unquenched LHCII intermediate, described here for the first time, provides insight into the molecular mechanism of quenching.
F. Li, C. Liu, S. Streckaite, C. Yang, P. Xu, M. J. Llansola-Portoles, C. Ilioaia, A. A. Pascal, R. Croce, and B. Robert.
A new, unquenched intermediate of LHCII. | Journal of Biological Chemistry, 2021 Jan 23.
Contact: Bruno ROBERT (bruno.robert@i2bc.paris-saclay.fr)




 We want to congratulate Dr. Denise Zickler for her election as an EMBO Member. Trained as a cytogeneticist, she has always been interested in the basic mechanisms that govern chromosome behavior during meiotic and mitotic divisions. Dr. Zickler has been a leader in the field since the 1980s and continues to push the community towards a better understanding of long-standing questions pertaining to homologous chromosome pairing and meiotic recombination. Her model system, the filamentous fungus Sordaria macrospora, became an unparalleled system to study meiosis. By clever genetic screens she identified all major players involved in the programmed DNA double-strand break formation that initiate meiotic recombination, ten to twenty years before their discovery in other organisms. 3D reconstruction of all seven chromosome pairs in each meiotic nucleus allowed to gain key insights into the pairing and recombination steps. This includes observation of the synaptonemal complex (SC), a proteinaceous structure between homologous chromosomes that stabilizes chromosome pairing and contributes to the regulation of crossover formation. By merging molecular, genetic and imaging analyses, Dr. Zickler has tackled the general problems of the connection between homologous pairing, SC initiation, crossover patterning with tremendous success (Cell, 2010; Genes Dev 2014; 2017; PNAS 2011; 2014). One of her last publications uncovered the existence of “bridges” between homologous chromosomes during the early stages of meiotic prophase. These bridges comprise structural components of the chromosome axes as well as recombination proteins. They allow to translocate the recombination proteins from the axes to the synaptonemal complex, mediating the interplay between this structure and the recombination process leading to crossover formation (PNAS 2019). She has been involved in many international collaborations, some that last to this day, bringing her intuition and her decisiveness to countless endeavors outside her own laboratory. Dr. Zickler always favored nurturing a small research group, wanting to stay nearly full time at the bench and believing in daily free exchanges of ideas. She has been an indispensable member of the meiosis community and, as such, has been an invited speaker at all major meiosis conferences for the last 20 years. She also spearheaded the creation of the EMBO Meiosis conference, that is still organized every other year and is a major event in the community. Her recognition as an EMBO member acknowledges her incomparable contribution over her fantastic career. Congratulations Denise!
We want to congratulate Dr. Denise Zickler for her election as an EMBO Member. Trained as a cytogeneticist, she has always been interested in the basic mechanisms that govern chromosome behavior during meiotic and mitotic divisions. Dr. Zickler has been a leader in the field since the 1980s and continues to push the community towards a better understanding of long-standing questions pertaining to homologous chromosome pairing and meiotic recombination. Her model system, the filamentous fungus Sordaria macrospora, became an unparalleled system to study meiosis. By clever genetic screens she identified all major players involved in the programmed DNA double-strand break formation that initiate meiotic recombination, ten to twenty years before their discovery in other organisms. 3D reconstruction of all seven chromosome pairs in each meiotic nucleus allowed to gain key insights into the pairing and recombination steps. This includes observation of the synaptonemal complex (SC), a proteinaceous structure between homologous chromosomes that stabilizes chromosome pairing and contributes to the regulation of crossover formation. By merging molecular, genetic and imaging analyses, Dr. Zickler has tackled the general problems of the connection between homologous pairing, SC initiation, crossover patterning with tremendous success (Cell, 2010; Genes Dev 2014; 2017; PNAS 2011; 2014). One of her last publications uncovered the existence of “bridges” between homologous chromosomes during the early stages of meiotic prophase. These bridges comprise structural components of the chromosome axes as well as recombination proteins. They allow to translocate the recombination proteins from the axes to the synaptonemal complex, mediating the interplay between this structure and the recombination process leading to crossover formation (PNAS 2019). She has been involved in many international collaborations, some that last to this day, bringing her intuition and her decisiveness to countless endeavors outside her own laboratory. Dr. Zickler always favored nurturing a small research group, wanting to stay nearly full time at the bench and believing in daily free exchanges of ideas. She has been an indispensable member of the meiosis community and, as such, has been an invited speaker at all major meiosis conferences for the last 20 years. She also spearheaded the creation of the EMBO Meiosis conference, that is still organized every other year and is a major event in the community. Her recognition as an EMBO member acknowledges her incomparable contribution over her fantastic career. Congratulations Denise!
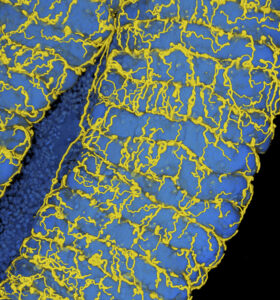 The insect respiratory system consists of tubular tracheae that transport oxygen to the organs. The tracheal network is dynamic and responds to developmental, environmental and nutritional cues. In a recent article published in the Proceedings of the National Academy of Sciences USA, the Plant Bacteria Interactions team of the Microbiology Department of I2BC, in collaboration with the team of Yoshitomo Kikuchi at the National Institute of Advanced Industrial Science and Technology – Hokkaido in Japan, shows that, in the insect pest Riptortus pedestris, the establishment of an essential symbiosis in the gut with the aerobic bacterial species Burkholderia insecticola triggers the development of an extensive tracheal network enveloping the gut. Genetically blocking the trachea formation prevents this gut symbiosis. The researchers further discovered that the reactive oxygen species-generating enzyme Duox is crucial for the formation and stabilization of tracheae by forming protein cross-links in the tracheal matrix. Reactive oxygen species generated by Duox can be scavenged with antioxidants such as N-acetylcysteine, and feeding insects with this compound prevents tracheal formation and symbiosis. Since many insects obligatorily depend on their symbioses, triggering their collapse by the specific inhibition of the respiratory network with antioxidants could be a new route to fight insect pests.
The insect respiratory system consists of tubular tracheae that transport oxygen to the organs. The tracheal network is dynamic and responds to developmental, environmental and nutritional cues. In a recent article published in the Proceedings of the National Academy of Sciences USA, the Plant Bacteria Interactions team of the Microbiology Department of I2BC, in collaboration with the team of Yoshitomo Kikuchi at the National Institute of Advanced Industrial Science and Technology – Hokkaido in Japan, shows that, in the insect pest Riptortus pedestris, the establishment of an essential symbiosis in the gut with the aerobic bacterial species Burkholderia insecticola triggers the development of an extensive tracheal network enveloping the gut. Genetically blocking the trachea formation prevents this gut symbiosis. The researchers further discovered that the reactive oxygen species-generating enzyme Duox is crucial for the formation and stabilization of tracheae by forming protein cross-links in the tracheal matrix. Reactive oxygen species generated by Duox can be scavenged with antioxidants such as N-acetylcysteine, and feeding insects with this compound prevents tracheal formation and symbiosis. Since many insects obligatorily depend on their symbioses, triggering their collapse by the specific inhibition of the respiratory network with antioxidants could be a new route to fight insect pests.
Ribosomes universally consist of two independent subunits that assemble on the mRNA during translation initiation. Surprisingly, it was shown that it was possible by synthetic biology to covalently link the two subunits together.
Although this is consistent with E. coli growth, the molecular consequences for translation fidelity were unknown. We therefore tested the functioning of this tethered ribosome, and were able to demonstrate that it is strongly impacted in its ability to recognise the SD sequence.
In consequence, initiation and SD-dependent frame shifting are both impacted. We were also able to show that the translation termination step was less efficient. It is precisely at this step that the two subunits are physically dissociated. The fact that this is no longer possible in the tethered ribosome clearly disrupts the release efficiency of the ribosome.
This work reveals the subtle perturbations linked to the attachment of the two ribosome subunits and should be taken into account in the biotechnological developments associated with the use of these tethered ribosomes.
Celine FABRET, Olivier NAMY, Translational accuracy of a tethered ribosome, Nucleic Acids Research, 2021
https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkab259/6266428#.YJLH79KSbnM.twitter

The Institute for Integrative Biology of the Cell (I2BC) is seeking to recruit junior and mid-career Group leaders to develop independent research programs in the fields of Virology and of Cell Biology.
Dead Line for application: april 15, 2021
The main research theme of I2BC concerns the characterization of the integrated functioning of the cell and in particular the understanding of the processes which, at the molecular level, govern the organization and the overall physiology of the cell. These studies, carried out in different model and non-model organisms (from viruses to human cells) aim to model life processes in order to integrate the impacts of molecular, subcellular and environmental changes in the physiology of normal or pathological cells and tissues.
I2BC wishes to reinforce the Departments of Virology and of Cell Biology. Candidates shall develop independent and high-quality fundamental research programs.
• Applications to the Department of Virology should cover the broad themes of structural and cell biology of virus infection. Research on virus entry, host-cell takeover, viral-induced cell compartmentalization, and cell intrinsic antiviral defenses are particularly encouraged.
• Applications to the Cell Biology Department are welcome on any aspect of cell biology, in particular in the biogenesis, organization, function and communication of cell structures and compartments, as well as their involvement in pathology or response to stress and infection.
Further information on scientific environment and application procedure can be found on the dedicated page : Call for new teams 2021