Summary
- Natural transformation and cell division delay in competent Staphylococcus aureus
- The large ring-shaped BRCA2-HSF2BP complex is disassembled by BRME1, thus promoting Homologous Recombination: a cryo-EM study.
- IP6 : an endogenous small molecule which promotes DNA repair.
- Functional and structural insights into the multi-step activation and catalytic mechanism of bacterial ExoY nucleotidyl cyclase toxins bound to actin-profilin
- The C-terminus of stathmin-like proteins governs the stability of their complexes with tubulin
- Discriminating Susceptibility of Xanthine Oxidoreductase Family to Metals
- Multiscale Transient Absorption Study of the Fluorescent Protein Dreiklang and Two Point Variants Provides Insight into Photoswitching and Nonproductive Reaction Pathways
- Clustered binding of the CTCF protein creates functional domains in the human and mouse genome with improved structure.
- Phosphorylation of polymerase theta by PLK1 is essential for the repair of double-strand breaks in mitosis through a novel DNA repair pathway.
- New virus/phage synteny web server: https://archaea.www.i2bc.paris-saclay.fr/vapex/
- How Rif1 bridles rapid genome replication during early developmental stages in vertebrae
- The universal Sua5/TsaC family evolved different mechanisms for the synthesis of a key tRNA modification
- The formation of structural domains in chromosomes: a highly dynamic process to create stable regulatory functions.
- Phosphatidylserine transport in cell life and death
- LGG-1/GABARAP lipidation is not required for autophagy and developmentin Caenorhabditis elegans
- Unexpected binding modes of ureabased foldamers to a protein surface
- How PAXX keeps DNA in check
- Archaeal conjugation at 100°C
- The Ccr4-Not complex is a major regulator of gene silencing and heterochromatin spreading
- Self assembling structures from artificial proteins, or how to design protein origamis.
- The complex regulation of competence in Staphylococcus aureus under microaerobic conditions
- Two essential complexes–Mediator and RSC chromatin remodeler–work together for chromatin organization at promoters
- Dissimilar gene repertoires of Dickeya solani involved in the colonization of lesions and roots of Solanum tuberosum
- Job offer
- AMMIB 2023
- An image-based high-content screening for compounds targeting Toxoplasma gondii repurposed inhibitors effective against the malaria parasite Plasmodium falciparum
- Dual-uptake mode of the antibiotic phazolicin by Gram-negative bacteria prevents resistance acquisition
- Manganese concentration affects chloroplast structure and the photosynthetic apparatus in Marchantia polymorpha
- Recipient chromosome-encoded UvrD helicase plays a role in plasmid acquisition by conjugationa
- A paralog of Pcc1 is the fifth core subunit of the KEOPS tRNA modifying complex in Archaea
- Rabies virus P protein binds to TBK1 kinase and interferes with the formation of innate immunity-related liquid condensates
- Convergent evolution of bacterial replicative helicase-helicase loader systems
- Exernal Seminar Jean Colcombet What are the missions for research facing energy-climate change crises ?
- Properties of rabies virus phosphoprotein and nucleoprotein biocondensates formed in vitro and in cellulo
- New insights into genome annotation in Podospora anserina through re‑exploiting multiple RNA‑seq data
- Strategic Axes Day
- How to tell a gene where and when it should be active?
- Accurate atomic model of the hepatitis E virus replication polyprotein
Straining the root on and off triggers local calcium signaling.
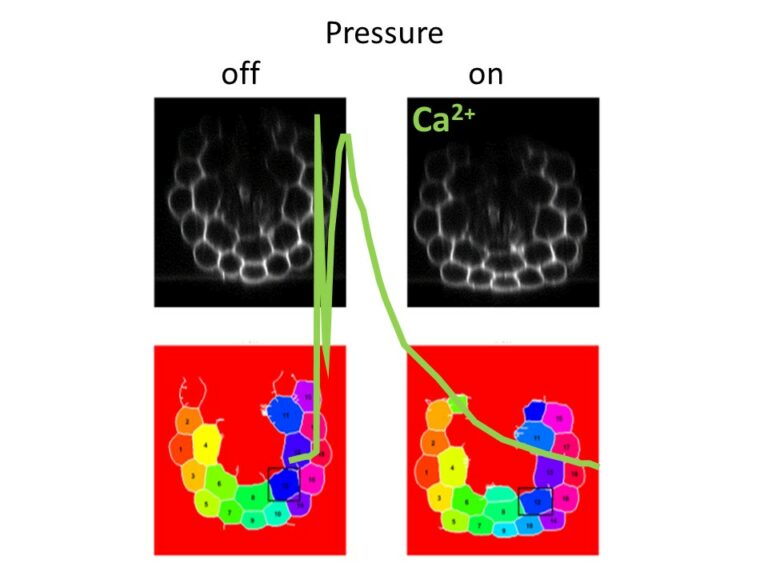
Combining microfluidics and imaging, reveal that a pressure exerted on the root induces an elastic deformation and a calcium signal both at the onset and at the offset of the stimulation.
How plant roots perceive mechanical cues encountered in the soil remains elusive. Combining microfluidics and imaging, we have shown that a pressure exerted on the root induces an elastic deformation and a calcium signal both at the onset and at the offset of the stimulation. Although the intensity of the calcium response increased with the pressure applied, successive stimuli led to a remarkable attenuation of the calcium signal. The calcium elevation was restricted to the tissue under pressure without propagation. This study published in Proceedings of the Royal Society B (https://doi.org/10.1098/rspb.2023.1462) contributes to elucidate the mechanisms of root adaptation to the mechanical cues generated by the soil.
This work was supported by the Région Ile de France through the DIM ELICIT program and by Saclay Plant Sciences through the DYNANO project.
More information: doi: https://doi.org/10.1098/rspb.2023.1462
Contact: Jean-Marie Frachisse – Sébastien Thomine <jean-marie.frachisse@i2bc.paris-saclay.fr> <sebastien.thomine@i2bc.paris-saclay.fr>
First anti-CRISPR protein that inhibits the CRISPR-Cas defense system in Clostridioides difficile
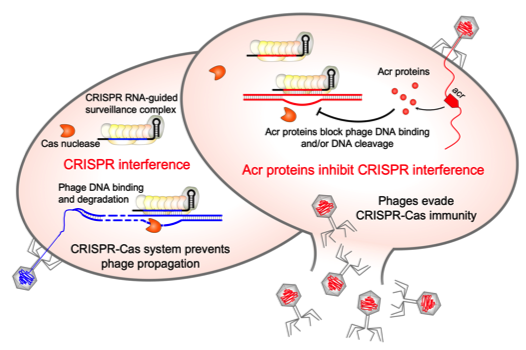
CRISPR-Cas adaptive immunity defends prokaryotes against their viruses named phages. In response, phages have evolved counter defense strategies to fight back. This study describes the identification of a phage protein to evade CRISPR-Cas immunity in a human pathogen Clostridioides difficile.
Clostridioides difficile is the widespread anaerobic spore-forming bacterium that is a major cause of potentially lethal nosocomial infections associated with antibiotic therapy worldwide. Due to the increase in severe forms associated with a strong inflammatory response and higher recurrence rates, a current imperative is to develop synergistic and alternative treatments of C. difficile infections. In particular, phage therapy is regarded as a potential substitute for existing antimicrobial treatments. However, it faces challenges because C. difficile have highly active CRISPR-Cas immunity, which may be a specific adaptation to phage-rich and highly crowded gut environment. To overcome this defense, C. difficile phages must employ anti-CRISPR mechanisms. Here, we present the first anti-CRISPR protein that inhibits the CRISPR-Cas defense system in this pathogen. Our work offers insights into the interactions between C. difficile and its phages, paving the way for future CRISPR-based applications and development of effective phage therapy strategies combined with the engineering of virulent C. difficile infecting phages.
More information: doi: 10.1128/msphere.00401-23
Contact: Olga SOUTOURINA <olga.soutourina@i2bc.paris-saclay.fr>
Inter-generational nuclear crosstalk controls the progression of the Paramecium developmental program
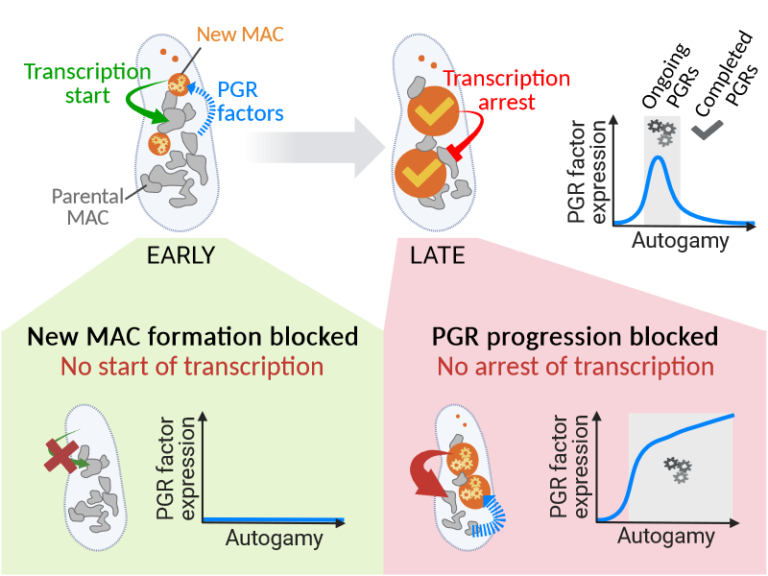
Perturbing the Paramecium transcriptome during somatic development reveals the existence of internuclear feed-forward and feedback transcriptional regulatory loops in a multinucleate unicellular eukaryote.
The single-celled ciliate Paramecium tetraurelia harbors multiple nuclei within its cytoplasm: two germline micronuclei (MIC) dedicated to sexual reproduction and a somatic macronucleus (MAC) responsible for gene expression. During the sexual cycle, the parental MAC undergoes progressive degradation while, in the same cell, new MACs develop from copies of the zygotic MIC. Throughout this process, gene expression primarily occurs in the parental MAC, and clusters of developmentally regulated genes are successively switched on and off. Concomitantly, the genome of the new MACs is extensively rearranged and cleansed of its transposons and other DNA repeats, to become competent for gene expression.
New MAC development has long been known to be controled by the parental MAC, which expresses key genes involved in programmed genome rearrangement (PGR). In this study, RNA deep sequencing and bioinformatic analysis of differential gene expression reveal a reciprocal relationship between the two generations of MACs: the formation of the new MACs is essential for the proper induction of a subset of developmental genes. Later in the sexual cycle, if PGR is hindered in the new MACs, a group of genes partially overlapping the former subset is upregulated, and no longer switched off. Thus, the progression of new MAC development regulates the gene expression program in the parental MAC at different stages. The set of co-expressed genes identified during the course of this work establishes a list of potential novel actors, whose roles in PGR will be explored in future studies.
More information: https://doi.org/10.1093/nar/gkad1006
Contact: Olivier Arnaiz – Mireille Bétermier <olivier.arnaiz@i2bc.paris-saclay.fr> <mireille.betermier@i2bc.paris-saclay.fr>
3D models of fungal chromosomes
to enhance visual integration of omics data
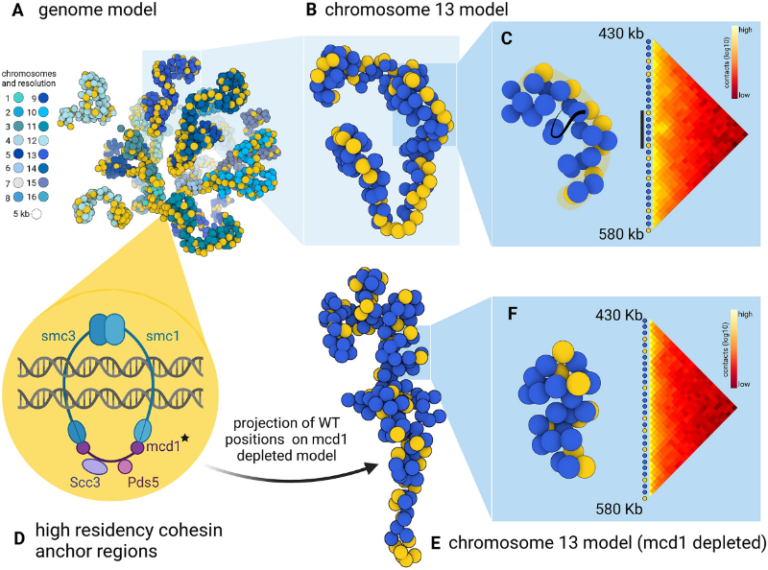
It has become easier to build 3D models of the entire genome based on Hi-C data, and thus benefit from the methodology and visualization tools developed for structural biology. In this paper, we show how seeing the spatial organization of chromosomes can bring new perspectives to omics data integration.
The functions of eukaryotic chromosomes and their spatial architecture in the nucleus are reciprocally dependent. Hi-C experiments are routinely used to study chromosome 3D organization by probing chromatin interactions. Standard representation of the data has relied on contact maps that show the frequency of interactions between parts of the genome. In parallel, it has become easier to build 3D models of the entire genome based on the same Hi-C data, and thus benefit from the methodology and visualization tools developed for structural biology. 3D modeling of entire genomes leverages the understanding of their spatial organization. However, this opportunity for original and insightful modeling is underexploited. In this paper, we show how seeing the spatial organization of chromosomes can bring new perspectives to omics data integration. We assembled state-of-the-art tools into a workflow that goes from Hi-C raw data to fully annotated 3D models and we re-analysed public omics datasets available for three fungal species. Besides the well-described properties of the spatial organization of their chromosomes (Rabl conformation, hypercoiling and chromosome territories), our results highlighted (i) in Saccharomyces cerevisiae, the backbones of the cohesin anchor regions, which were aligned all along the chromosomes, (ii) in Schizosaccharomyces pombe, the oscillations of the coiling of chromosome arms throughout the cell cycle and (iii) in Neurospora crassa, the massive relocalization of histone marks in mutants of heterochromatin regulators. 3D modeling of the chromosomes brings new opportunities for visual integration of omics data. This holistic perspective supports intuition and lays the foundation for building new concepts.
More information: https://academic.oup.com/nargab/article/5/4/lqad104/7458894
Contact: Gaëlle LELANDAIS <gaelle.lelandais@i2bc.paris-saclay.fr>
CryoEM studies of the human lipid transporter ATP8B1 involved in intrahepatic cholestasis

The structural characterization of the catalytic cycle of the lipid transporter ATP8B1 by cryoEM reveals fundamental aspects of its substrate specificity and of its regulation.
Lipids are the main constituents of our cell membranes, which are formed as lipid bilayers. The distribution of lipids is far from uniform; it is asymmetric, with different lipid compositions in the inner and outer leaflet. This asymmetry is essential for a variety of cellular functions, from maintaining membrane homeostasis to enabling cell signaling and numerous other physiological processes including apoptosis and coagulation.
P4-ATPases, also known as flippases, are key players in creating and maintaining this lipid asymmetry. These enzymes actively transport lipids from the outside (exoplasmic) leaflet to the inside (cytosolic) leaflet coupled to ATP hydrolysis and ensure the proper distributions of lipids. The ATP8B1-CDC50A flippase complex in particular, has been the subject of the current study, is involved in a rare inherited liver disease called intrahepatic cholestasis. The function of ATP8B1 lipid flippase is critical for the regulation of bile production, a vital substance in our digestive system, but the direct link within bile producing liver cells remains unknown. Additionally, recent studies have spotlighted the relevance of genetic variants in the regulatory segment of the ATP8B1 gene as a strong genetic marker for Alzheimer’s resilience.
In the new study, the research team employed state-of-the-art cryo-electron microscopy techniques to capture nine different states associated with the lipid transport and determine structures at 2.4 to 3.1 Å overall resolution for these conformations. These structural insights, combined with functional and computational studies, reveal the inner workings of the human flippase ATP8B1-CDC50A complex and also resolves earlier discrepancies about the ATP8B1 transport substrates. Additionally, this work also provides key understanding about fine regulation by specific regulatory lipids known as phosphoinositides and by its autoinhibition mechanism.
This work was recently awarded ”article of the month” by the Société Française de Biochimie et Biologie Moléculaire (SFBBM) in December 2023.
More information: https://www.nature.com/articles/s41467-023-42828-9
Contact: Thibaud DIEUDONNE <thibaud.dieudonne@i2bc.paris-saclay.fr>
Natural transformation and cell division delay in competent Staphylococcus aureus
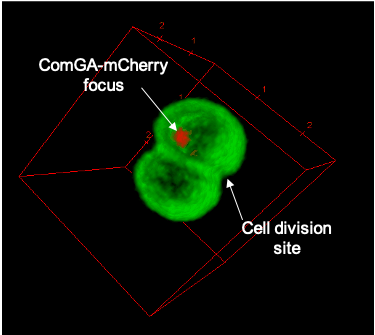
We hypothesize that S. aureus competent cells would initiate and then block cell division to ensure the success of natural transformation before the final constriction of the cytokinetic ring..
Genetic competence for natural transformation, considered one of the three main mechanisms leading to horizontal gene transfer in bacteria, is able to promote evolution, through genomic plasticity, and foster antibiotic resistance and virulence factors spreading. Conserved machinery and actors required to perform natural transformation have been shown to accumulate at different cellular localizations depending on the model organism considered. In this study, we investigate the transformation apparatus composition, localization, and dynamics in the human pathogen Staphylococcus aureus. We particularly show that most of the natural transformation actors co-localize in clusters. We also reveal that the localization of natural transformation proteins is dynamic, following the cell cycle. Ultimately, the natural transformation apparatus is preferentially established in the vicinity of the division septum. All these results demonstrate that DNA binding, uptake, and recombination are spatially and temporally coordinated to ensure S. aureus natural transformation. Finally, we hypothesize that S. aureus competent cells would initiate and then block cell division to ensure the success of natural transformation before the final constriction of the cytokinetic ring.
More information: https://journals.asm.org/doi/10.1128/spectrum.02807-23
Contact: Nicolas MIROUZE <nicolas.mirouze@i2bc.paris-saclay.fr>
The large ring-shaped BRCA2-HSF2BP complex is disassembled by BRME1, thus promoting Homologous Recombination: a cryo-EM study.
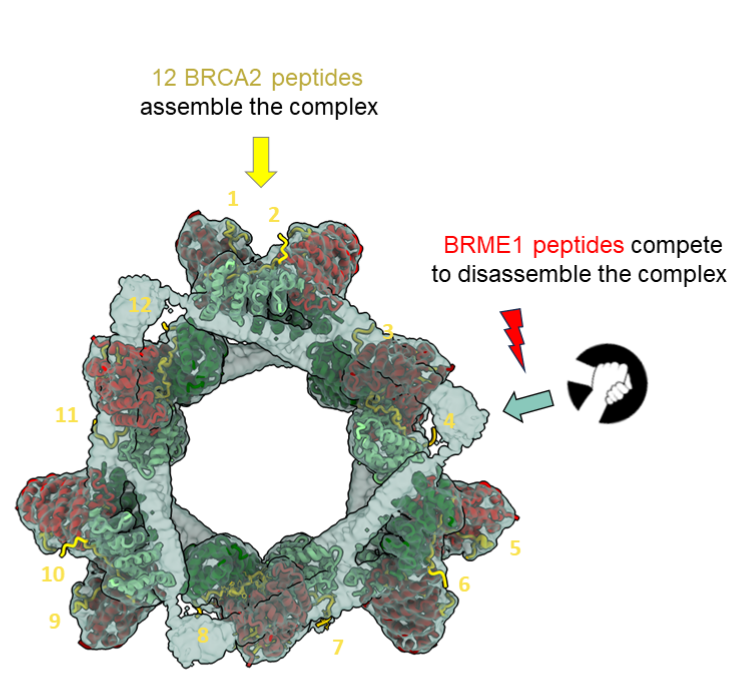
A short disordered and conserved motif of the tumor suppressor BRCA2 triggers the assembly of a large ring-shaped 24-mer of the meiotic HSF2BP protein, whereas the disordered C-terminal motif of the meiotic BRME1 dissociates this ring complex.
In meiotic homologous recombination (HR), BRCA2 facilitates loading of the recombinases RAD51 and DMC1 at the sites of double-strand breaks (DSB). The HSF2BP-BRME1 complex interacts with BRCA2. Its absence causes a severe reduction in recombinase loading at meiotic DSB. We previously showed that, in somatic cancer cells ectopically producing HSF2BP, DNA damage can trigger HSF2BP-dependent degradation of BRCA2, which prevents HR. Here we report that, upon binding to BRCA2, HSF2BP forms octameric rings that are able to assemble into a large ring-shaped 24-mer. Addition of BRME1 leads to dissociation of both of these ring structures, and cancels the disruptive effect of HSF2BP on cancer cell resistance to DNA damage. It also prevents BRCA2 degradation during inter-strand DNA crosslink repair in Xenopus egg extracts. We propose that the control of HSF2BP-BRCA2 oligomerization by BRME1 ensures timely assembly of the ring complex that concentrates BRCA2 and controls its turnover, thus promoting HR.
More information: https://www.science.org/doi/10.1126/sciadv.adi7352
Contact: Sophie ZINN JUSTIN <sophie.zinn@i2bc.paris-saclay.fr>
IP6 : an endogenous small molecule which promotes DNA repair.
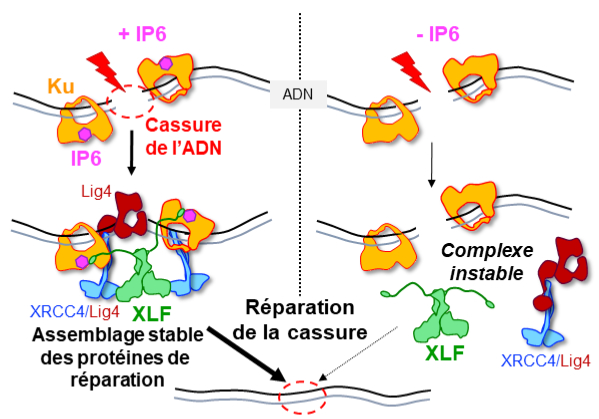
Structural studies by X-ray cristallography and CryoEM combined with cellular analyses show the stabilizing role of IP6 in assembly of DNA double-strand break repair in human.
IP6, inositol hexaphosphate or phytic acid, is a product of cellular metabolism that binds to numerous proteins whose functions it regulates. In an article published in the journal Nucleic Acids Research, scientists from I2BC (UMR 9198, CNRS/CEA/UPSaclay, Gif-sur-Yvette), from lPBS (CNRS/Université Paul Sabatier) and from Institut for Structural and Chemical Biology (University of Leicester, UK) reveal how IP6 stabilizes the assembly of a complex responsible for DNA break repair in humans.
Many cancer therapies induce DNA double-strand breaks. A DNA double-strand break represents the most serious type of DNA damage to a cell, since it amounts to splitting a chromosome in two. The resulting chromosome fragments can be lost or lead to translocations if they are not quickly rejoined. Unrepaired breaks most often lead to cell death, a property exploited to eradicate tumor cells during radiotherapy or certain chemotherapies. Yet DNA double-strand break repair pathways exist, and their performance in tumors determines the efficacy of these therapies. The dominant system for repairing DNA double-strand breaks in human cells is Non-Homologous End Joining, or NHEJ, initiated by the Ku protein, which rapidly encircles the ends of the break and acts as a hub for the other proteins needed to weld these ends together.
Using two techniques for studying proteins at atomic scale (crystallography and cryo-electron microscopy), the scientists discovered how a small molecule produced by the body and sometimes used as a dietary supplement, inositol-hexaphosphate (IP6) or phytic acid, binds to the Ku protein.
The scientists then analyzed the effect of IP6 on break repair in human cells. They modified the Ku protein to block its binding to IP6, and thus understand how the latter acts. The Ku mutants revealed that the presence of IP6 on the Ku protein stimulates its binding to the XLF protein, an essential step in efficiently welding the DNA break (see the model below). IP6 thus plays a key role in stabilizing the large complex of NHEJ proteins required for DNA break repair. This fundamental work resolves the question raised two decades ago regarding the role of IP6 in DNA double-strand break repair. It also opens up therapeutic prospects by identifying a new region on the Ku protein that could be targeted by small molecules to block DNA break repair in tumor cells.
More information: https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkad863/7327077?login=false
Contact: Jean-Baptiste CHARBONNIER <jb.charbonnier@i2bc.paris-saclay.fr>
Functional and structural insights into the multi-step activation and catalytic mechanism of bacterial ExoY nucleotidyl cyclase toxins bound to actin-profilin
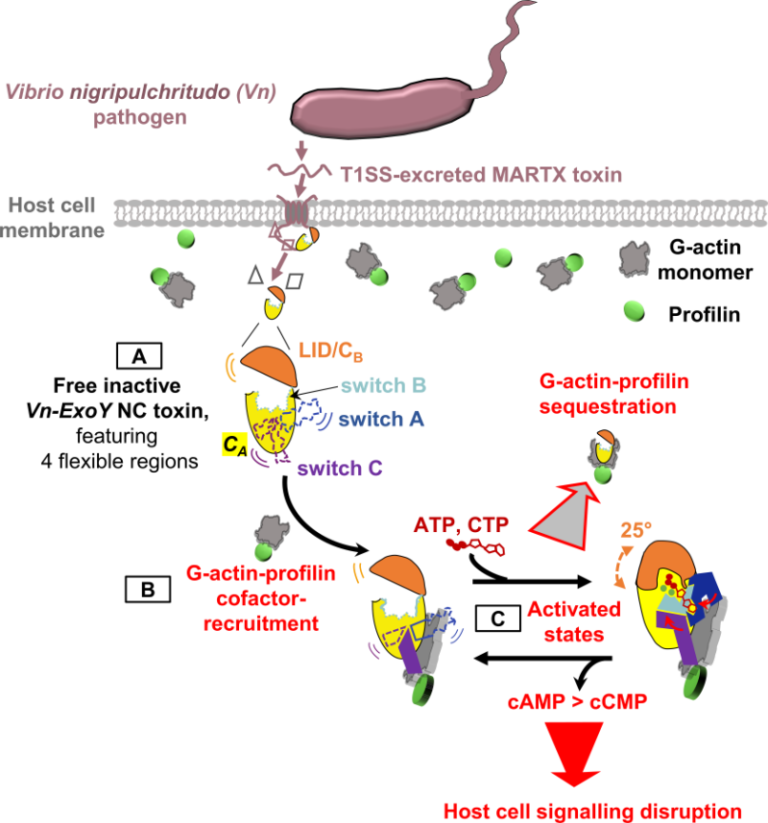
We reveal unprecedented mechanistic structural details of how the active site of bacterial Nucleotidyl Cyclase toxins is sequentially remodeled by cofactor and substrate binding and how they can accommodate both purine or pyrimidine nucleotides as substrate.
Nucleotidyl cyclase (NC) enzymes are important ubiquitous enzymes that catalyze the production of cyclic nucleotides, crucial second messengers involved in numerous signaling pathways. ExoY-like toxins are bacterial virulence factors produced by Gram-negative γ- and β-proteobacteria. They belong to a broader family of bacterial NC toxins. When delivered into eukaryotic cells, NC toxins alter host cell signalling by overproducing both cyclic purine (cAMP, cGMP) and pyrimidine (cCMP, cUMP) nucleotides. To become potent NC enzymes inside host cells, they bind to specific host cofactors. The molecular and mechanistic details underlying the activation and catalytic specificities of NC toxins are only partially understood. ExoY NC toxins utilize either polymerized or monomeric (G-actin) actin as cofactor.
Here, we (a team at I2BC collaborating with scientists at ICSN, CNRS, UPS, Gif-Sur-Yvette, and at Institute Pasteur, Paris) investigated ExoY-like NCs that are selective for G-actin. Our in vitro investigations first revealed the physiological cofactor at work within host eukaryotic cells. These ExoYs activate by capturing and sequestering the cytoskeletal G-actin-profilin complex, disrupting its regulatory functions in the dynamic assembly of actin (Fig. 1, steps B-C).
Using X-ray crystallography at SOLEIL synchrotron, we have intercepted several structural snapshots along the ExoY activation pathway by G-actin-profilin. These structural data reveal unprecedented mechanistic details of how the active site of all NC toxins undergoes progressive remodeling upon cofactor and substrate binding (Fig. 1, step C). They reveal how NC toxins can accommodate purine or pyrimidine nucleotides as substrates and clarify crucial aspects of their catalytic purinyl and pyrimidinyl cyclase reaction. These structural insights into the multi-step activation of NC toxins may open new avenues to inhibit specifically this class of toxic bacterial NCs.
More information: https://journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1011654
Contact: Louis RENAULT <louis.renault@i2bc.paris-saclay.fr>
The C-terminus of stathmin-like proteins governs the stability of their complexes with tubulin
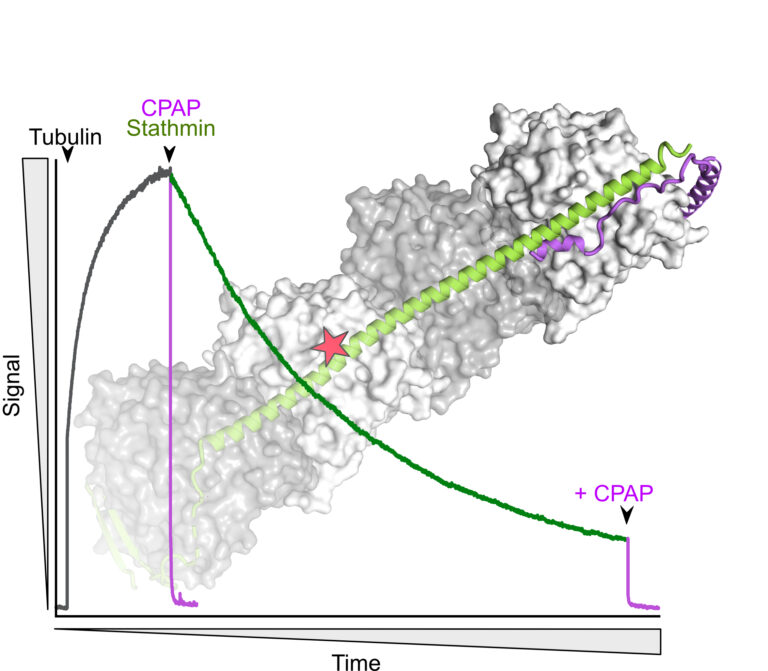
Identification of a novel mechanism for the regulation of the stathmin:tubulin interaction.
Microtubule dynamics is modulated by many cellular factors including stathmin family proteins. Vertebrate stathmins sequester two ab-tubulin heterodimers into a tight complex that cannot be incorporated in microtubules. Stathmins are regulated at the expression level during development and among tissues; they are also regulated by phosphorylation. We have studied the dissociation kinetics of tubulin:stathmin assemblies in presence of different tubulin-binding proteins and identified a critical role of the C-terminus of the stathmin partner. Destabilizing this C-terminal region may represent an additional regulatory mechanism of the interaction with tubulin of stathmin proteins.
More information: https://doi.org/10.1016/j.bbrc.2023.10.023
Contact: Benoît GIGANT <benoit.gigant@i2bc.paris-saclay.fr>
Discriminating Susceptibility of Xanthine Oxidoreductase Family to Metals
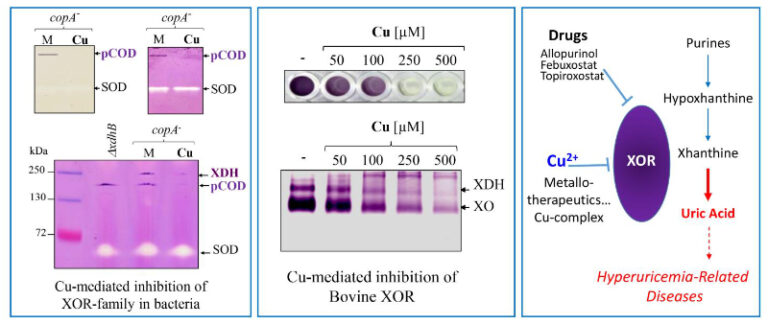
Copper as an inhibitor of the Xanthine Oxidoreductase family in purple bacteria and mammals, highlighting its potential use in metal-based therapeutics against hyperuricemia and gout arthritis.
The xanthine oxidoreductases (XORs) family are metal-containing enzymes that use the molybdenum cofactor (Moco), 2Fe-2S clusters, and Flavin adenine dinucleotide (FAD) for their catalytic activity. This large molybdoenzymes family includes xanthine, aldehyde and CO-dehydrogenases. XORs are widely distributed from bacteria to humans due to their key roles in the catabolism of purines, aldehydes, drugs, and xenobiotics, as well as interconversions between CO and CO2. Assessing the effect of excess metals on Rubrivivax gelatinosus bacterium, we found that exposure to copper (Cu) or cadmium (Cd) caused a dramatic decrease in the activity of a high molecular weight soluble complex exhibiting nitroblue-tetrazolium reductase activity. Mass spectrometry and genetic analyses showed that the complex corresponds to a putative CO-dehydrogenase (pCOD). Using mutants that accumulate either Cu+ or Cd2+ in the cytoplasm, we show that Cu+ or Cd2+ are potent inhibitors of XORs (pCOD and the xanthine dehydrogenase) in vivo. This is the first in vivo demonstration that Cu+ affects Moco containing enzymes. The specific inhibitory effect of these compounds on the XORs activity is further supported in vitro by direct addition of competing metals to protein extracts. Moreover, emphasis is given on the inhibiting effect of Cu on Bovine XOR, showing that the XORs family could be a common target of Cu. Given the conservation of XORs structure and function across the tree of life, we anticipate that our findings could be transferable to other XORs and organisms.
More information: https://doi.org/10.1128/spectrum.04814-22
Contact: Soufian OUCHANE <soufian.ouchane@i2bc.paris-saclay.fr>
Multiscale Transient Absorption Study of the Fluorescent Protein Dreiklang
and Two Point Variants Provides Insight into Photoswitching and Nonproductive Reaction Pathways

Photoswitching and non-productive reaction pathways of the fluorescent protein ‘Dreiklang’ deciphered in a comprehensive time-resolved spectroscopic study.
Dreiklang is a reversibly photoswitchable fluorescent protein used as a probe in advanced fluorescence imaging. It undergoes a unique and still poorly understood photoswitching mechanism based on the reversible addition of a water molecule to the chromophore. We report the first comprehensive study of the dynamics of this reaction by transient absorption spectroscopy from 100 fs to seconds in the original Dreiklang protein and its two point variants. The picture that emerges from our work is that of a competition between photoswitching and nonproductive reaction pathways. We found that photoswitching had a low quantum yield of 0.4%. It involves electron transfer from a tyrosine residue (Tyr203) to the chromophore and is completed in 33 ns. Nonproductive deactivation pathways comprise recombination of a charge transfer intermediate, excited-state proton transfer from the chromophore to a histidine residue (His145), and decay to the ground state via micro-/millisecond-lived intermediates.
More information: https://pubs.acs.org/doi/10.1021/acs.jpclett.3c00431
Contact: Pavel MULLER <pavel.muller@i2bc.paris-saclay.fr>
Identification of a new pathway potentially involved in plant resistance to drought
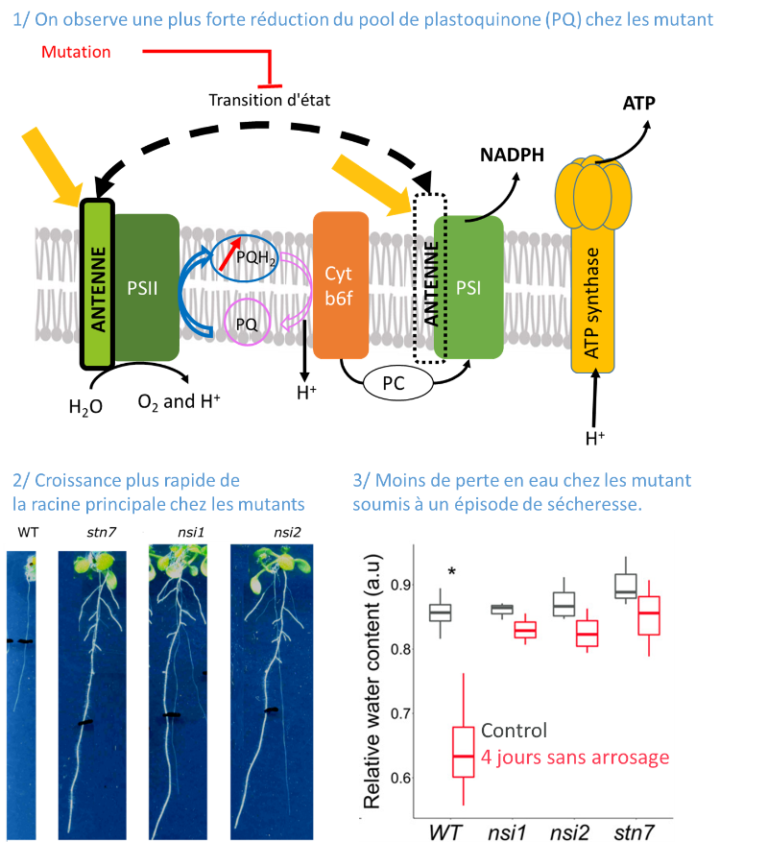
Light capture by the chlorophyll containing antenna and the reduction state of the photosynthetic electron transport chain affect root development.
Identifying traits that exhibit improved drought resistance is highly important to cope with the challenges of predicted climate change. We investigated the response of state transition mutants to drought. Compared with the wild type, state transition mutants were less affected by drought. Photosynthetic parameters in leaves probed by chlorophyll fluorescence confirmed that mutants possess a more reduced plastoquinone (PQ) pool, as expected due to the absence of state transitions. Seedlings of the mutants showed an enhanced growth of the primary root and more lateral root formation. The photosystem II inhibitor 3-(3,4-dichlorophenyl)-1,1-dimethylurea, leading to an oxidised PQ pool, inhibited primary root growth in wild type and mutants, while the cytochrome b6 f complex inhibitor 2,5-dibromo-3-methyl-6-isopropylbenzoquinone, leading to a reduced PQ pool, stimulated root growth. A more reduced state of the PQ pool was associated with a slight but significant increase in singlet oxygen production. Singlet oxygen may trigger a, yet unknown, signalling cascade promoting root growth. We propose that photosynthetic mutants with a deregulated ratio of photosystem II to photosystem I activity can provide a novel path for improving crop drought resistance.
More information: https://pubmed.ncbi.nlm.nih.gov/37614199/
Contact: Anja KRIEGER-LISZKAY <anja.krieger-liszkay@i2bc.paris-saclay.fr>
Clustered binding of the CTCF protein creates functional domains in the human and mouse genome with improved structure.
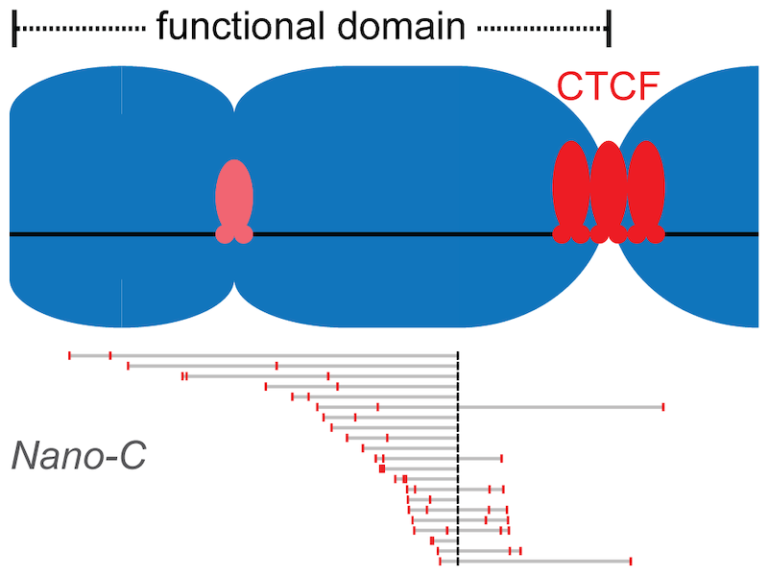
Clusters of binding sites for the CTCF protein create boundaries between functional domains in the mouse and human genomes. Scientists at the I2BC have determined the characteristics of these clusters and how individual sites contribute to domain separation.
Topologically Associating Domains (TADs) regionalize the human and mouse genome into domains that are essential for processes like gene regulation and DNA repair. Most boundaries between TADs bind the CTCF insulator protein, which is responsible for the blocking of interactions between neighboring TADs. CTCF is thereby directly responsible for improved functional precision and reduced regulatory interference between domains in the genome. Although a clustering of CTCF binding sites had previously been observed at TAD boundaries, its structural impact remained to be determined.
Scientists in the Chromatin Dynamics group at the I2BC, in collaboration with groups at the Ecole Normale Supérieure (Paris) and the University of Pennsylvania (USA), found that three features of CTCF binding are enriched at TAD boundaries, including the presence of closely-spaced copies of its DNA binding motifs, a reduced distance between clusters of motifs and an enrichment of clusters with improved binding affinity. Using Nano-C technology, a new approach to measure the blocking of DNA interactions, they subsequently determined the blocking capacity of individual sites of CTCF binding. These studies reveal that these sites individually contribute to the blocking of interactions between domains, but in an incomplete manner. Grouping of multiple sites of CTCF binding in the genome thus creates a stepwise insulation between neighboring TADs, thereby improving the focus of genomic functions.
More information: https://doi.org/10.1038/s41467-023-41265-y
Contact: Daan NOORDERMEER <daan.noordermeer@i2bc.paris-saclay.fr>
Phosphorylation of polymerase theta by PLK1 is essential for the repair of double-strand breaks in mitosis through a novel DNA repair pathway.
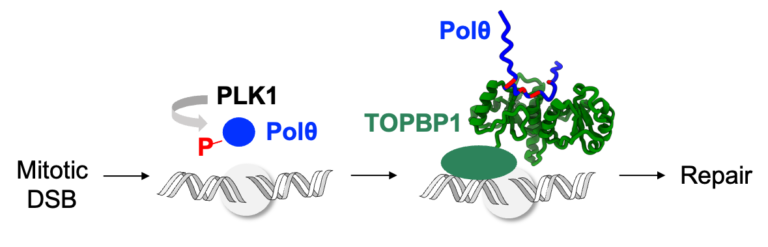
The DNA polymerase theta is phosphorylated by PLK1 in mitosis, binds to TOPBP1 and is recruited to double-strand breaks, in order to trigger repair through a novel DNA repair pathway, thus becoming a new target for the treatment of breast and ovarian cancers.
DNA double-strand breaks (DSBs) are deleterious lesions that challenge genome integrity. In interphase, DSBs are mainly repaired by non-homologous end joining and homologous recombination. In mitosis, specific kinases inhibit these pathways through phosphorylation of essential DNA repair proteins. Here we show that one of these mitotic kinases, Polo-like kinase 1 (PLK1), activates the DNA polymerase theta (Polθ), which is then recruited through an interaction with TOPBP1 to mitotic DSBs. NMR analyses demonstrate that PLK1 phosphorylates a cluster of four serines in the central disordered region of Polθ, and that the phosphorylated motif directly interacts with the C-terminal domains of TOPBP1. The Artificial Intelligence based program AlphaFold consistently predicts that the Polθ region containing the cluster of four serines binds in a groove at the surface of the C-terminal domains of TOPBP1. Mutating these serines impairs recruitment of Polθ to mitotic DSBs and joining of the broken DNA ends. Polθ is essential for the repair of mitotic DSBs. Its role is even more crucial in cells that are deficient in homologous recombination, because these cells accumulate DSBs at the entry of mitosis, and loss of mitotic DSB repair by Polθ results in cell death. Our data explains why Polθ is synthetic lethal with homologous recombination deficiency, and reveals the critical importance of mitotic DSB repair in the maintenance of genome integrity.
More information: https://www.nature.com/articles/s41586-023-06506-6
Contact: Sophie ZINN <sophie.zinn@i2bc.paris-saclay.fr>
New virus/phage synteny web server:
https://archaea.www.i2bc.paris-saclay.fr/vapex/
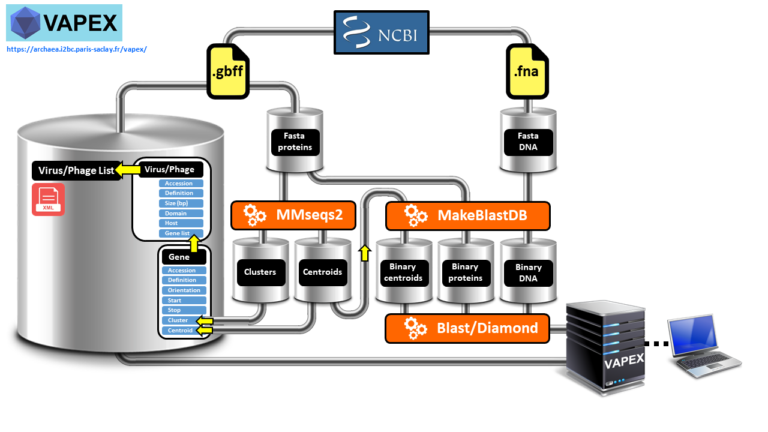
VAPEX creates fully resolved synteny maps of all natural and user-submitted virus and phage genomes.
Studying the genetic makeup of viruses and phages through genome analysis is crucial for comprehending their function in causing diseases, progressing medicine, tracing their evolutionary history, monitoring the environment, and creating innovative biotechnologies. However, accessing the necessary data can be challenging due to a lack of dedicated comparative genomic tools and viral and phage databases, which are often outdated. Moreover, many wet bench experimentalists may not have the computational proficiency required to manipulate large amounts of genomic data.We have developed VAPEX (Virus And Phage EXplorer), a web server which is supported by a database and features a user-friendly web interface. This tool enables users to easily perform various genomic analysis queries on all natural viruses and phages that have been fully sequenced and are listed in the NCBI compendium. VAPEX therefore excels in producing visual depictions of fully resolved synteny maps, which is one of its key strengths. VAPEX has the ability to exhibit a vast array of orthologous gene classes simultaneously through the use of symbolic representation. Additionally, VAPEX can fully analyze user-submitted viral and phage genomes, including those that have not yet been annotated.
More information: https://doi.org/10.1093/bioinformatics/btad528
Contact: Jacques Oberto <jacques.oberto@i2bc.paris-saclay.fr>
How Rif1 bridles rapid genome replication during early developmental stages in vertebrae
Combined experimental and in-sillico approaches show that the Rif1 protein restricts the activation of replication origins of chromatin domains and the recruitment of initiation factors during the first embryonic divisions.
In multicellular eukaryotic organisms, genomic DNA replicates in a time-controlled manner by ordering early, intermediate, or late replication regions according to a spatio-temporal replication program. How this program is orchestrated is poorly understood, but its dysregulation leads to genomic instability often observed in cancer. The Rif1 protein is a key regulator of this program in eukaryotes; however, its role during the first embryonic cell cycles, when DNA replication is very rapid and replication factors are abundant, remained poorly characterized. Researchers from I2BC, NeuroPSI, ENS Paris, and CalTech (USA) have clarified these mechanisms using the high-performance in vitro system of Xenopus egg extracts by combining the analysis of DNA fibers by molecular combing with an in-sillico model. This study, published in Communications Biology, reveals that in the absence of Rif1, the temporal program of replication is greatly accelerated at the level of groups of origins. This acceleration is accompanied by increased chromatin recruitment of an S phase kinase (Cdc7/Drf1) and several other key factors implicated in the replication initiation (Treslin/MTBP, RecQL4). The model proposed in this study is that Rif1 simultaneously restricts access to DNA or the activity of several factors to fine-tune the exceptionally rapid DNA synthesis observed during embryonic development. A better understanding of the role of Rif1 during these stages opens a way to elucidating the molecular mechanisms involved in certain diseases resulting from Rif1 mutations or variants in humans.
Read on to find out more in Nature Portfolio: https://cellmolbiocommunity.springernature.com/posts/how-rif1-bridles-rapid-embryonic-dna-synthesis
Contact: Kathrin Marheineke <kathrin.marheineke@i2bc.paris-saclay.fr>
The universal Sua5/TsaC family evolved different mechanisms
for the synthesis of a key tRNA modification
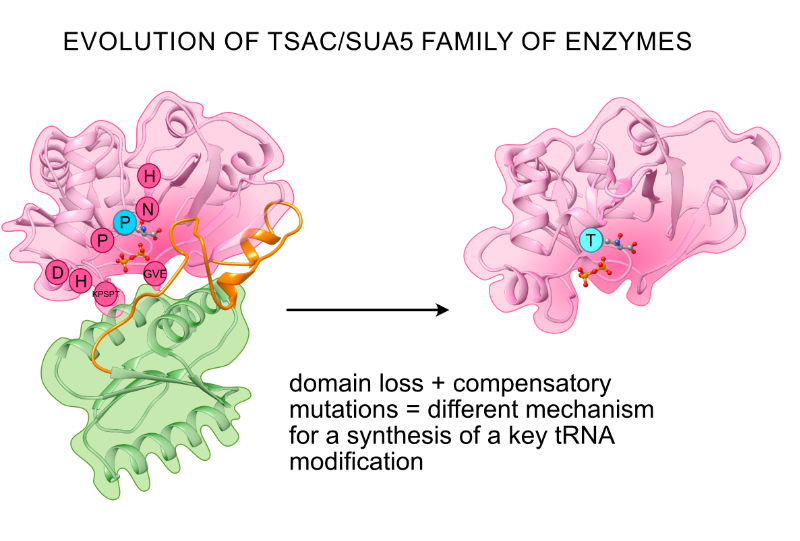
TsaC/Sua5 family of enzymes catalyzes the first step in the synthesis of N6-threonylcarbamoyl adenosine (t6A) one of few truly ubiquitous tRNA modifications important for translation accuracy. TsaC is a single domain protein while Sua5 proteins contains a TsaC-like domain and an additional SUA5 domain of unknown function. The emergence of these two proteins and their respective mechanisms for t6A synthesis remain poorly understood. Here, we performed phylogenetic and comparative sequence and structure analysis of TsaC and Sua5 proteins. We confirm that this family is ubiquitous but the co-occurrence of both variants in the same organism is rare and unstable. We further find that obligate symbionts are the only organisms lacking sua5 or tsaC genes. The data suggest that Sua5 was the ancestral version of the enzyme while TsaC arose via loss of the SUA5 domain that occurred multiple times in course of evolution. Multiple losses of one of the two variants in combination with horizontal gene transfers along a large range of phylogenetic distances explains the present day patchy distribution of Sua5 and TsaC. The loss of the SUA5 domain triggered adaptive mutations affecting the substrate binding in TsaC proteins. Finally, we identified atypical Sua5 proteins in Archaeoglobi archaea that seem to be in the process of losing the SUA5 domain through progressive gene erosion. Together, our study uncovers the evolutionary path for emergence of these homologous isofunctional enzymes and lays the groundwork for future experimental studies on the function of TsaC/Sua5 proteins in maintaining faithful translation.
More information: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1204045/full
Contact: Tamara Basta-Le Berre <tamara.basta@i2bc.paris-saclay.fr>
The formation of structural domains in chromosomes:
a highly dynamic process to create stable regulatory functions.
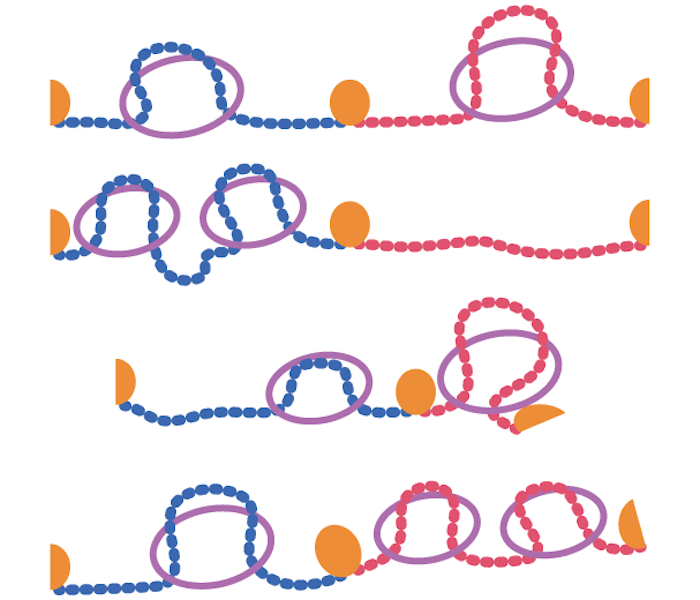
Our chromosomes are divided into strucrural domains that focus biological activity. In this literature review, researchers from the I2BC summarize recent insights into the dynamic nature of this process, and how this nonetheless can create stable regulation.
Mammalian chromosomes are organized at different length scales within the cell nucleus. Topologically Associating Domains (TADs) are structural units of 3D genome organization that compartmentalize chromosomes into separated domains. Within these domains, biological functions are focused, thereby restricting the “spread” of gene regulation, DNA replication, recombination and repair. In this review, published in Current Opinion in Structural Biology, scientists from the Chromatin Dynamics team at the I2BC discuss recent insights into the structure and function of TADs. Whereas TADs were initially interpreted as insulated domains, recent studies are revealing that these domains should be interpreted as dynamic collections of actively extruding loops. This process of loop extrusion is subsequently blocked at dedicated TAD boundaries, thereby promoting intra-domain interactions over their surroundings. The authors discuss how mammalian TAD structure can emerge from this dynamic process and they discuss recent evidence that boundaries between TADs can have regulatory functions.
More information: https://www.sciencedirect.com/science/article/abs/pii/S0959440X23000969
Contact: Daan Noordermeer <dann.noordermeer@i2bc.paris-saclay.fr>
Phosphatidylserine transport in cell life and death
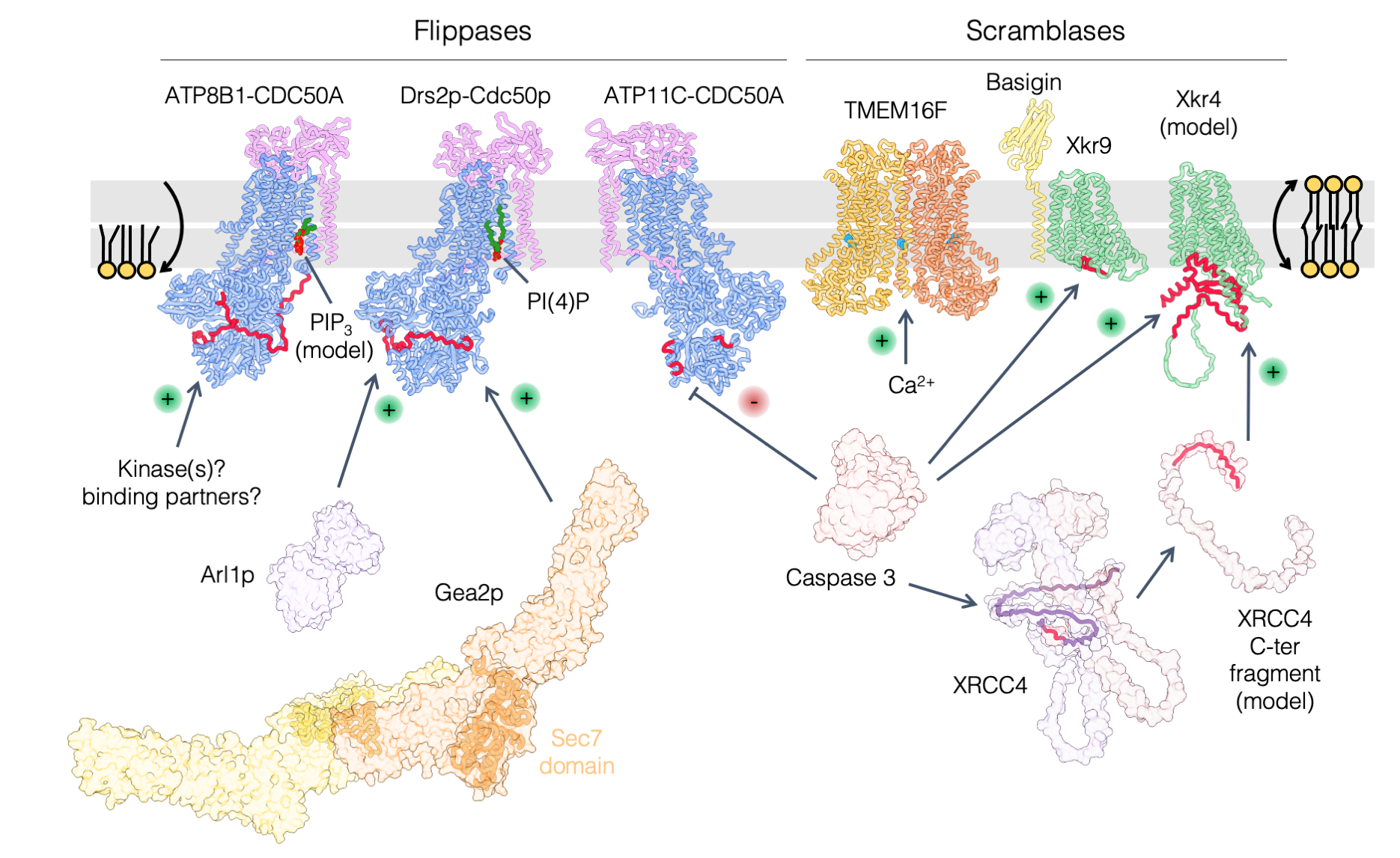
Lipids beyond membrane building blocks: recent advances in our understanding of phosphatidylserine distribution in eukaryotic cells and its role in infectious diseases.
A prominent feature of cell membranes is the non-uniform distribution of the lipids they are made of. This is particularly true for the negatively charged glycerophospholipid phosphatidylserine (PS) which is enriched in the plasma membrane and in late secretory/endocytic compartments. In addition to the uneven distribution of PS between membranes, the transbilayer distribution (the enrichment of a given lipid in one membrane leaflet over the other) of PS is tightly regulated, as it is largely confined to the cytosolic leaflet of membranes, where it can mediate the recruitment of a diverse set of proteins. As such, PS controls a myriad of cell signaling pathways as well as membrane trafficking events. In some specific conditions, PS may however be ‘flipped’ toward the external leaflet of the plasma membrane, where it can act as an ‘eat-me’ signal for engulfment of apoptotic cells by macrophages or to promote synaptic pruning, which consists in the removal of synapses to establish proper connections during brain development. The exquisitely tailored transbilayer PS distribution is also crucial in host-pathogen interactions, as it is exploited by various intracellular pathogens to infect cells. In this review, we highlight recent findings on nonvesicular PS transport between membranes by lipid-transfer proteins at membrane contact sites, on PS flip-flop between membrane leaflets by lipid flippases and scramblases, and we discuss how perturbation of PS distribution can lead to disease and the role of PS in viral infection.
More information: https://www.sciencedirect.com/science/article/pii/S0955067423000418?via%3Dihub
Contact: Guillaume Lenoir <guillaume.lenoir@i2bc.paris-saclay.fr>
LGG-1/GABARAP lipidation is not required for autophagy and development
in Caenorhabditis elegans
Using a CRISPR approach in C. elegans, this study shows that the ubiquitin-like ATG8 homolog LGG-1 possesses lipidation-independent function in autophagy, and provides a novel insight into the role of ATG family during animal development.
The ubiquitin-like proteins Atg8/LC3/GABARAP are required for multiple steps of autophagy, such as initiation, cargo recognition and engulfment, vesicle closure and degradation. Most of LC3/GABARAP functions are considered dependent on their post-translational modifications and their association with the autophagosome membrane through a conjugation to a lipid, the phosphatidyl-ethanolamine. Contrarily to mammals, C. elegans possesses single homologs of LC3 and GABARAP families, named LGG-2 and LGG-1. Using site-directed mutagenesis, we inhibited the conjugation of LGG-1 to the autophagosome membrane and generated mutants that express only cytosolic forms, either the precursor or the cleaved protein. LGG-1 is an essential gene for autophagy and development in C. elegans, but we discovered that its functions could be fully achieved independently of its localization to the membrane. This study reveals an essential role for the cleaved form of LGG-1 in autophagy but also in an autophagy-independent embryonic function. Our data question the use of lipidated GABARAP/LC3 as the main marker of autophagic flux and highlight the high plasticity of autophagy.
More information: https://elifesciences.org/articles/85748
Contact: Renaud Legouis <renaud.legouis@i2bc.paris-saclay.fr>
Unexpected binding modes of ureabased foldamers to a protein surface
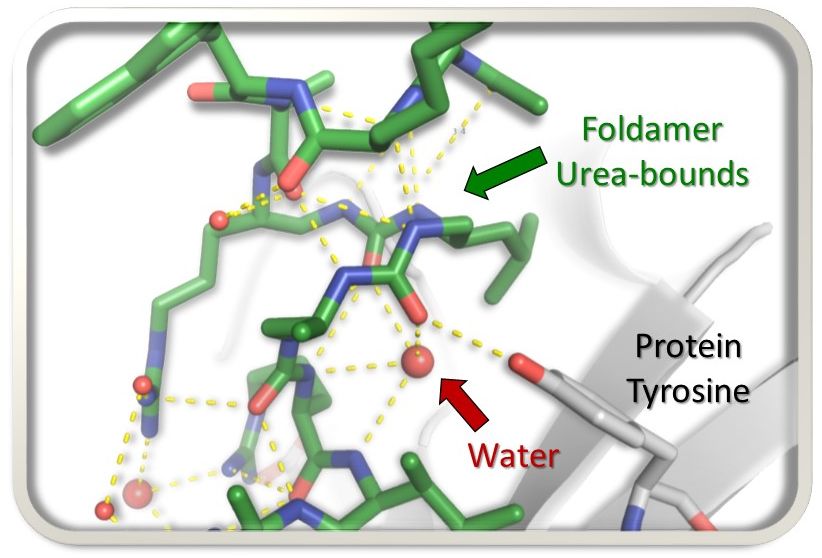
Exploring uncharted territory, peptide-urea chimeras display surprising binding modes to the histone chaperone ASF1, rewriting the rules of interaction.
In the search for foldamer inhibitors of the histone chaperone ASF1, the AMIG team of I2BC in collaboration with a team of Bordeaux University, explored the possibility of substituting four α-residues (≈ one helix turn) by 3-urea segments and scanned the sequence of a short α-helical peptide known to bind ASF1. By analysing the impact of the different foldamer replacements within the peptide chain, new binding modes of the peptide-urea chimeras to a protein target were discovered.
More information: https://doi.org/10.1039/D3CC01891A
Contact: Françoise Ochsenbein <francoise.ochsenbein@i2bc.paris-saclay.fr>
How PAXX keeps DNA in check

We visualized the crucial steps of the molecular assembly allowing cells to repair breaks in their DNA, in particular those involving the PAXX protein.
« It’s in my DNA » fine, but the information has to stay there without too much change, at the risk of losing our identity! Monitoring the health of DNA is the job of the many repair systems in our cells, each specialized in detecting and correcting a specific anomaly. A particularly serious threat is the breakage of the DNA double chain. For the cell, this is like a train whose tracks are suddenly interrupted: serious damage ahead! But the DNA is a fragile molecule that breaks easily, for example during flight under the effect of cosmic radiation, or upon exposure with radioactivity or X-rays, or simply because cells grow under reactive oxygen. The rail maintenance department in human cells is called the NHEJ, which coordinates many agents to detect breaks, protect them by a safety cord and finally weld them together. Of all the agents involved, the PAXX protein was the latest identified in 2015 but its role was not fully known. A B3S team in collaboration with a IPBS team in Toulouse (P Calsou) and a UK team in Bristol (A Chaplin) succeeded in reconstructing how PAXX functions in NHEJ. When the DNA is broken, repair begins with the ring-like Ku protein that very quickly encircles the ends of the break, serving as a mooring platform for the other proteins. Using two techniques that allow proteins to be visualized at the atomic scale (X-rays crystallography and cryo-electron microscopy), we captured a snapshot of the moment when PAXX docks with Ku and precisely identified their contact zones. They showed that PAXX works in tandem with XLF attached to the other side of Ku. When the binding sites are disrupted by mutations, the whole machinery is blocked, the break is no longer repaired, the train derails and the cell dies! In hyperactive cancer cells, the train travels at high-speed, explaining their special sensitivity to DNA breaks induced by radiotherapy ; molecules that would block the assembly of PAXX and/or XLF on DNA breaks could make it possible to increase the vulnerability of cancer cells and thus open up new therapeutic perspectives. The results of this research are therefore very promising and could lead to more effective therapies for cancer patients, although there is still work to be done to find and test these molecules.
More information: https://pubmed.ncbi.nlm.nih.gov/37256950/
Contact: Jean-Baptiste Charbonnier <jb.charbonnier@i2bc.paris-saclay.fr> – Virginie Ropars <virginie.ropars@i2bc.paris-saclay.fr>
Archaeal conjugation at 100°C

Double world record: first experimental report of archaeal conjugation & first experimental evidence of conjugation at 100°C.
Conjugative plasmids are self-transmissible mobile genetic elements which transfer DNA between host cells via Type IV Secretion Systems (T4SS). While T4SS-mediated conjugation has been well-studied in diderm bacteria, information from monoderm bacteria remains sparse, and even less is known for the Archaea, largely due to their limited genetic accessibility. To date, only one family of conjugative plasmids has been described in Archaea, with all representatives coming from the Sulfolobales order of Crenarchaeota. Here we present the first self-transmissible plasmid identified in a Euryarchaeon, Thermococcus sp. 33-3. The 103 kbp plasmid, pT33-3, has left evidence of its past transfer in CRISPR-spacers throughout the Thermococcales order, and encodes virB4/trbE and virD4/traG homologues characteristic of T4SS. Using genetic techniques, we demonstrate that pT33-3 is a bona fide conjugative plasmid, with transmissibility being plasmid-encoded, requiring cell-to-cell contact, and dependent upon proteins constituting canonical T4SS. pT33-3 transfers under laboratory conditions to various Thermococcales, and transconjugants propagate at 100°C. Using pT33-3, we developed a genetic toolkit which allows modification of phylogenetically diverse Archaeal genomes. We demonstrate pT33-3–mediated plasmid mobilization and subsequent targeted genome modification in previously untransformable Thermococcales species, and extend this process to interphylum transfer to a Crenarchaeon.
More information: https://www.nature.com/articles/s41564-023-01387-x
Contact: Jacques Oberto <jacques.oberto@i2bc.paris-saclay.fr>
The Ccr4-Not complex is a major regulator of gene silencing and heterochromatin spreading
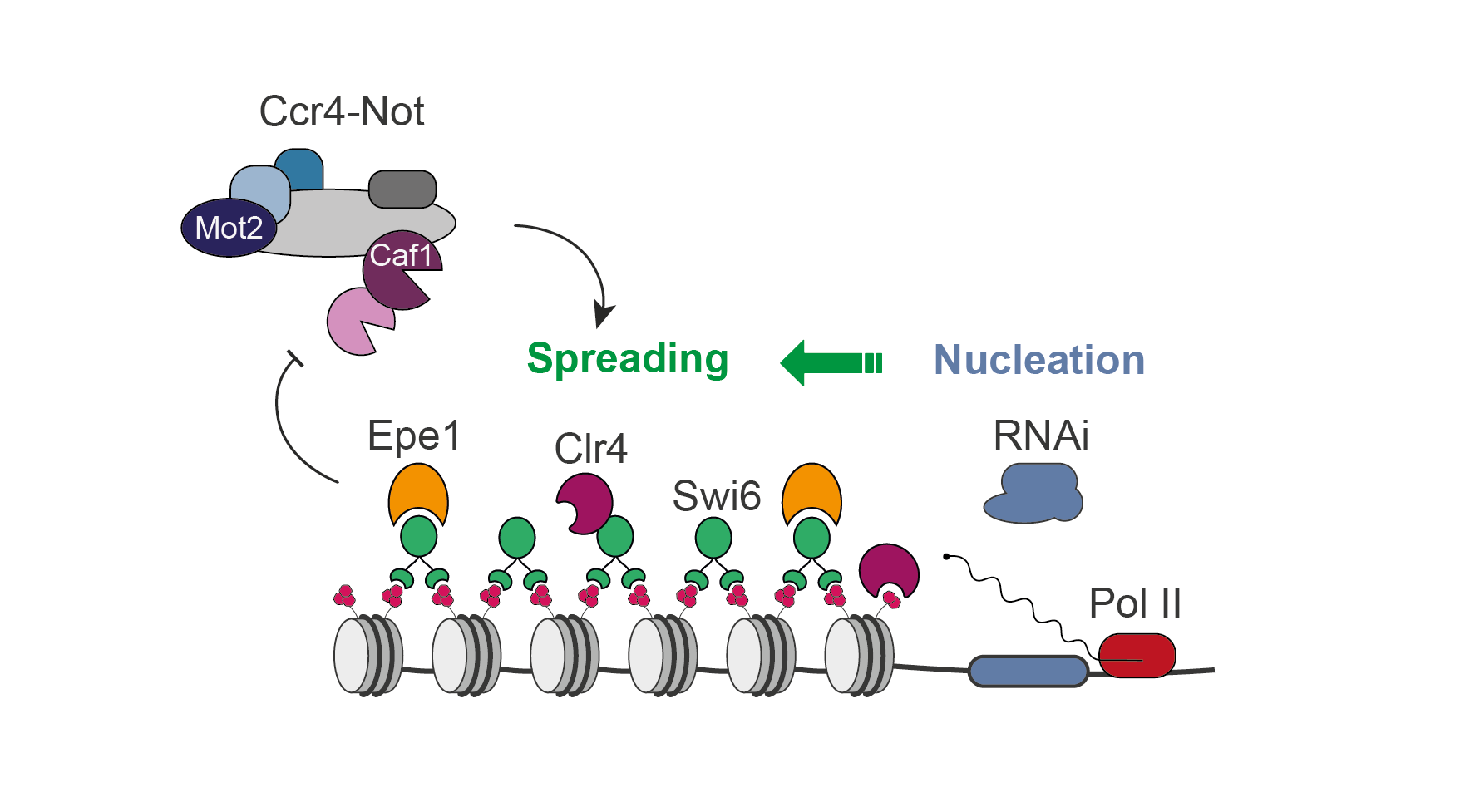
The fission yeast Ccr4-Not complex promotes propagation of the repressive H3K9me3 histone mark to mediate heterochromatic gene silencing.
Eukaryotic genomes are partitioned into relaxed, gene-rich regions and condensed, gene-poor domains called heterochromatin. The maintenance of heterochromatin is crucial for proper genome expression and integrity, and requires multiple factors regulating histone modifications and/or the levels of RNA molecules produced from these regions. Such effectors not only promote heterochromatin assembly but also ensure its propagation from specific nucleation sites to defined domain boundaries. However, while the mechanisms involved in initiation of heterochromatin formation have been well documented, the molecular and biochemical properties underlying its spreading remain largely elusive. By combining genetic and single-cell approaches, we report here that the fission yeast Ccr4-Not complex, a multisubunit complex conserved throughout eukaryotes, is essential for efficient heterochromatin spreading to repress expression of nucleation-distal RNAs. The two catalytic activities of the complex, RNA deadenylation and protein ubiquitinylation, are each critical, thereby defining a dual enzymatic requirement in the process.
More information: https://academic.oup.com/genetics/advance-article/doi/10.1093/genetics/iyad108/7190671?login=false
Contact: Mathieu Rougemaille <mathieu.rougemaille@i2bc.paris-saclay.fr>
Self assembling structures from artificial proteins, or how to design protein origamis.
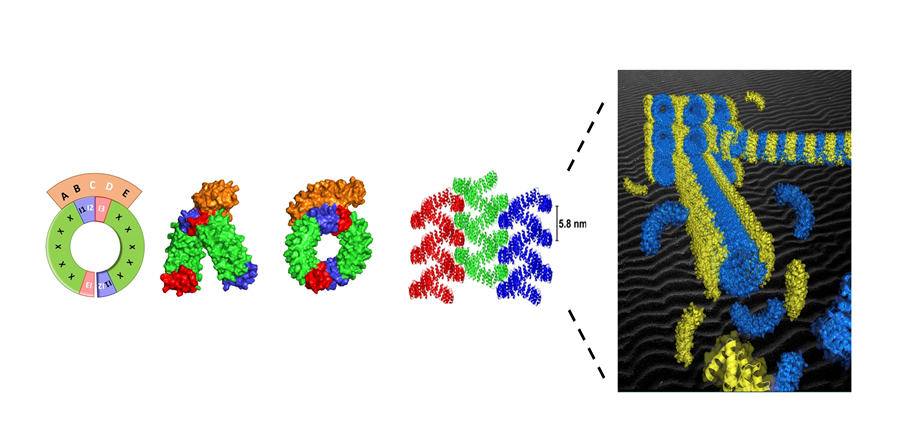
Organized superstructures made from proteins such as microtubules, cilia or flagella are ubiquitous in living cells. How to design such highly organized superstructure from artificial protein components ?
We describe a versatile strategy to create inducible protein assembly with predefined geometry. The assembly is mediated by a binding protein that staples identical protein bricks together in a predictable geometry. The first step was to select, among the alphaRep protein library, a protein able to interact with a specific area (the “back” surface) of another repeat protein used as a Bait. The structure of the “Bait”/”Back-binder” complex was then solved by the I2BC protein crystallography facility. Based on this structure, we designed a new protein, named the “Brick”, in which the surface elements of the Bait protein interacting with the “Back-binder” were split in two parts and appended at the two extremities of the “Brick” protein. This is made possible due to the extremely stable and modular organization of repeats proteins, such as alphaRep. In presence of the Brick protein, the Back-binder reconstitutes its cognate binding surface by joining the C end of one Brick protein to the N end of another Brick protein, in such a way that the surface elements of the two linked bricks adopt the same relative orientation as in the bait protein. Due to the twisted solenoid shape of the brick protein, the concatenation of many brick proteins leads to the formation of micrometer long super-helical filaments with a highly regular geometry. A very detailed structural characterization of the resulting assembly was conducted together with the Group of Erik Dujardin (CEMES Toulouse) using Negative stain electron microscopy (I2BC electron microscopy facility) , Small angle X ray scattering (F. Artzner, CNRS Rennes) , Cryoelectron Microscopy and Tomography (Integrative electron microscopy , Université de Toulouse). These efforts result in a detailed experimental description of the molecular organization and explain the higher order organization of the filaments by interdigitation of the back binders from neighboring filaments. The principles demonstrated here are potentially general and can be transferred to other types of affinity protein pairs derived either from natural or from computationally designed protein pairs and open routes toward the design and fabrication of multiscale protein origami with arbitrarily programmed shapes and chemical functions…
More information: https://www.pnas.org/doi/10.1073/pnas.2218428120
Contact: Philippe Minard <philippe.minard@i2bc.paris-saclay.fr>
The complex regulation of competence in Staphylococcus aureus under microaerobic conditions
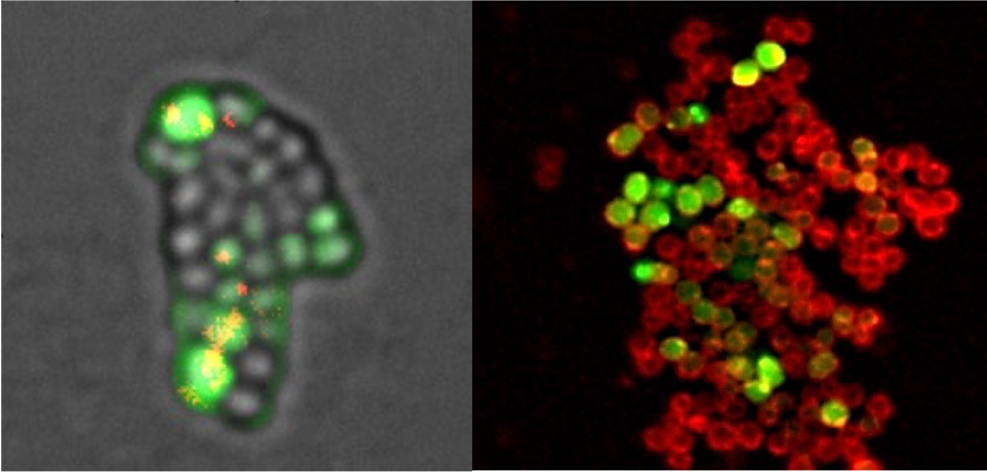
Where, when and how does Staphylococcus aureus acquire new antibiotic resistance genes ?
According to the World Health Organization (WHO), antimicrobial resistance (AMR) is one of the top 10 global public health threats facing humanity. In Europe, the health burden of antibiotic resistant bacteria is comparable to that of influenza, tuberculosis and HIV/AIDS combined and can be considered as a “hidden pandemic”.
Horizontal Gene Transfer (HGT), which can be defined as the transfer of genetic sequences between organisms with no parent-offspring relationship, is essential for the acquisition of new antibiotic resistance genes. Natural Transformation, one of the three main HGT mechanisms in bacteria, ensures the binding, internalization and homologous recombination within the host chromosome of exogenous sequences present in the environment. Importantly, in order to perform natural transformation, bacteria need to enter a differentiated state, called Genetic Competence. Many human pathogenic bacteria have the ability to induce genetic competence for natural transformation. Importantly, the human pathogen Staphylococcus aureus ability to induce competence have recently been characterized.
In this study, we designed a new protocol proving S. aureus ability to very efficiently induce competence and transformation. Taking advantage of this protocol, we characterized the three central competence regulators, all essential but playing different roles. Finally, we demonstrated that oxygen rarefaction was an important environmental stress for the induction of competence. This last result is not trivial considering that S. aureus encounters such microaerobic conditions during infection.
More information: https://www.nature.com/articles/s42003-023-04892-1
Contact: Nicolas Mirouze <nicolas.mirouze@i2bc.paris-saclay.fr>

Two essential complexes--Mediator and RSC chromatin remodeler--work together for chromatin organization at promoters
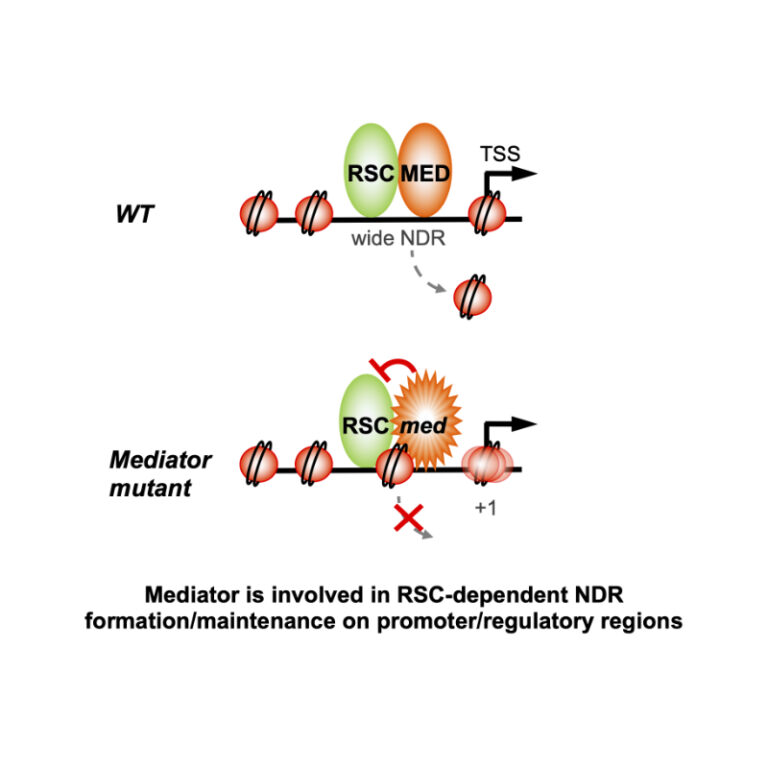
Physical interaction and functional interplay between essential co-regulators in transcription and chromatin organization, Mediator and RSC chromatin remodeler, contributes to nucleosome-depleted region formation at promoters and +1 nucleosome positioning.
Eukaryotic DNA is compactly structured through chromatin organization which governs and influences all DNA transactions. Regulation of transcription is a key phenomenon of gene expression whose alterations lead to severe human pathologies. Multisubunit coregulator complexes can act on chromatin structure as chromatin modifiers or remodelers, or stimulate the assembly of the transcriptional machinery. Mediator is an essential and conserved coactivator thought to act in concert with chromatin regulators. However, it remains largely unknown how their functions are coordinated. The team of J. Soutourina (Genome biology/I2BC and Institute Joliot, CEA/CNRS/Paris-Saclay) provides evidence in the budding yeast that Mediator establishes physical contact with RSC (Remodels the Structure of Chromatin), a conserved and essential chromatin remodeling complex that is crucial for nucleosome-depleted region (NDR) formation. The role of Mediator-RSC interaction in their chromatin binding, nucleosome occupancy and transcription was determined on a genomic scale. Mediator and RSC co-localize on wide NDRs of promoter regions, and specific Mediator mutations affect nucleosome eviction and stability of +1 nucleosome associated with transcription-start site. This work shows that Mediator contributes to RSC remodeling function to shape NDRs and maintain chromatin organization on promoter regions. It will help in our understanding of transcriptional regulation in the chromatin context relevant for severe diseases.
More information: doi.org/10.1016/j.celrep.2023.112465
Contact: Julie Soutourina <julie.soutourina@i2bc.paris-saclay.fr>
Dissimilar gene repertoires of Dickeya solani involved in the colonization of lesions and roots of Solanum tuberosum

High Throughput Screening Reveals genes involved in pre-symptomatic colonization of potato roots by a bacterial pathogen.
Dickeya and Pectobacterium species are necrotrophic pathogens that macerate stems (blackleg disease) and tubers (soft rot disease) of Solanum tuberosum. They proliferate by exploiting plant cell remains. They also colonize roots, even if no symptoms are observed. The genes involved in pre-symptomatic root colonization are poorly understood. Here, transposon-sequencing (Tn-seq) analysis of Dickeya solani living in macerated tissues revealed 126 genes important for competitive colonization of tuber lesions and 207 for stem lesions, including 96 genes common to both conditions. In root colonization, Tn-seq highlighted 83 genes, all different from those in stem and tuber lesion conditions. This work revealed novel traits and pathways important for understanding how the D. solani pathogen efficiently survives on roots, persists in the environment, and colonizes progeny tubers.
More information: https://doi.org/10.3389/fpls.2023.1154110
Contact: Denis Faure <denis.faure@i2bc.paris-saclay.fr>

JOB DESCRIPTION
Teaching
– Training fields concerned: The Junior Professor will participate in the development of a training program in structural biochemistry integrating the latest methodological developments in this field. He/she will notably contribute to the evolution of the specialized courses of the Master Health-Biology at Paris-Saclay in order to follow the evolution of the challenges in Structural Biochemistry and its applications in the current research. His/her teachings could also be addressed to the students of the international course proposed in the Master Health-Biology and contribute to the international influence of the University Paris-Saclay through the European partnership Eugloh. He/she will also participate the Life Sciences bachelor degree program, through the teaching of fundamental biochemistry concepts and in particular the structure of biological macromolecules, in order to attract students to Structural Biochemistry and ensure the future of research in this field.
– Teaching objectives and supervision requirements: The teaching duties of the recruited person will have to integrate electron microscopy technologies in the broad sense, as for example isolated single particle cryoEM analysis for atomic scales and imaging techniques such as tomography for more mesoscopic scales. These courses should also include the new modeling tools that have recently made it possible, thanks to artificial intelligence, to predict the structure of many proteins.
– Team leadership and participation in the life of the institution: The Junior Professor will be involved in the reflection on the renewal of the Master Health-Biology. He/she will propose the necessary evolutions, notably in the teaching of the different techniques of structural biology to the students of the course “Biomolecular Engineering and Chemistry” (M1 &M2).
Research activities
– Research project in the host laboratory: Within the Institute for Integrative Biology of the Cell (I2BC), the department of “Biochemistry, Biophysics and Structural Biology” (B3S) brings together 13 research teams whose themes are centered on the multiscale structural study, from the atom to multicellular organisms, of biological macromolecules and their assemblies, particularly in the field of health. Membrane proteins represent 60% of therapeutic targets and are the subject of innovative strategies such as immunotherapy. They play an essential role in the development of infectious diseases. The Junior Professor will have to propose a research project in line with the desire of the B3S department to play a major role in the development of structural studies of membrane proteins. His/her expertise in production and biochemical characterization of membrane complexes as well as in cryo-EM will enable him/her to overcome the methodological obstacles inherent to this theme and will open up new perspectives in the understanding of cellular processes controlled by membrane proteins. The upcoming construction of a P3 laboratory at the I2BC will allow the Junior Professor to develop an interdisciplinary project focused on membrane proteins involved in host/pathogen interactions. As the host/microbe theme is one of the 4 transversal research axes of the institute, the Junior Professor would find in the rich scientific environment of the I2BC the necessary collaborations to broaden his/her research towards studies at the cell and/or organism level.
– Research objectives in relation with the strategy of the university: In the field of Life Sciences, the University of Paris-Saclay is particularly interested in the structure-function analysis of biological macromolecules and the presence of the SOLEIL synchrotron, associated with numerous research teams in Structural Biology, is at the heart of the influence of the University of Paris-Saclay in this field. Over the last ten years, this field has evolved considerably thanks to the progress made in cryo-electron microscopy (cryo-EM). A significant effort has been made on the Université Paris-Saclay campus in the development of this technology with the installation of two latest generation electron microscopes at SOLEIL and at the I2BC. Through the Graduate School “Life Science and Health”, and in particular the interdisciplinary “LivingMachines@work” project, the University of Paris-Saclay is supporting this transformation. In order to maintain a high level of excellence in this field, the Graduate Program “Biochemistry & Structural Biology”, and in particular the “Engineering and Chemistry of Biomolecules” course of the Master in Biology and Health, must integrate these technological evolutions in their teaching. The recruitment of a Junior Professor in structural biochemistry specialized in cryo-EM will allow the University of Paris-Saclay to position itself at the forefront of this field, both in research and in training.
– Leadership in research animation : It is expected that the recruited person has a strong potential to lead a research group, as well as to participate in national, European or international projects. Candidates must demonstrate their ability to develop an innovative scientific project that can strengthen one of the transverse axes of the I2BC.
– Open science: An open science approach is expected. The results must be deposited in the open science archive HAL (https://hal.archives-ouvertes.fr/) or bioRxiv (https://www.biorxiv.org/). The structural data will be deposited in free open data banks, such as the PDB (Protein data Bank, https://www.ebi.ac.uk/pdbe/), the EMDB (Electron Microscopy Data Bank https://www.ebi.ac.uk/emdb/) and the EMPIAR (Electron Microscopy Public Image Archive https://www.ebi.ac.uk/empiar/).
Any other element related to the future research mission: The laureate of the Chair will be awarded a 200k€ grant by the french ANR (of which 150k€ will be for salaries – doctoral students, post-docs, technician/ingineer contractual staff- the balance being reserved for the project’s operation). The I2BC will also provide a 50k€ installation package and the B3S department will also pay 50k€ for access to the I2BC platforms.
Contact: olivier.lespinet@i2bc.paris-saclay.fr & sylvie.nessler@i2bc.paris-saclay.fr
Register via GALAXIE:
https://galaxie.enseignementsup-recherche.gouv.fr/antares/can/astree/index.jsp
4th meeting of the GdR MéDynA, October 3-6, 2023

registration open until May 15, 2023: https://www.azur-colloque.fr/DR04/inscription/inscription/258
Self-assembled molecular systems are studied in various fields ranging from physics to biology, including chemistry, mathematics and materials science. While the properties (mechanical, biological) of complex molecular assemblies are already the subject of well-established scientific fields, the study of the dynamic phenomena leading to their formation and evolution over time remains a field in genesis. However, this is not only a field of fundamental scientific interest, but also of practical interest, such as for pathological assemblies or assemblies with therapeutic or (bio)technological purposes. For more details on MéDynA : https://medyna.cnrs.fr/
More information: here
Symposium "Environment and Host Microbiomes - Functional Interactions"

Bringing together researchers working on fundamental, translational and clinical research in the field of “Microbiota”.
The symposium is organized by the GS LSH and the OI MICROBES on Wednesday, May 31, 2023 at Henri Moissan, and aims to bring together researchers from different fields (basic research, translational and clinical research) working on the topic of “Microbiota”. The objective of the symposium is to provide the opportunity to the different communities to know each other and to initiate collaborations in the future.
Registration is free but mandatory: https://www.universite-paris-saclay.fr/actualites/environment-and-host-microbiomes-functional-interactions
Young researchers (PhD students and Post-Doc) are invited to present their research as a poster (abstract to be submitted before May 5, 2023 online: https://cirrus.universite-paris-saclay.fr/s/58tPW6HpnS2Fbz8)
Contact: Peter MERGAERT <peter.mergaert@i2bc.paris-saclay.fr>
Stéphane Abel
 We are particularly sad to announce the death of our colleague Stéphane Abel, which occurred on Monday March 13, 2023.
We are particularly sad to announce the death of our colleague Stéphane Abel, which occurred on Monday March 13, 2023.
Since 2003, Stéphane worked with M. Marchi in B. Robert’s team, first during his PhD, then as a Marie Curie post-doc. He was recruited by the CEA in 2008 as a researcher, obtained his “Habilitation à Diriger des Recherches” at Université Paris-Saclay in 2017, and was recently promoted to “Directeur de Recherches” by the CEA.
S. Abel was interested in multi-scale modelling and molecular dynamics simulations of biological processes. He developed and used atomistic and coarse-grained molecular dynamics to study the stability and function of membrane proteins in different environments (normal phase or reverse micelles, or phospholipid membranes). He also used quantum mechanical approaches to develop force field parameters for biomolecules and, in particular, the detergents used in membrane protein experiments.
In addition to his qualities as a researcher, Stéphane was an extremely endearing character. This little man suffered from a serious brittle bone disease, and his life was undoubtedly a long struggle – but Stéphane was never bitter. He had a sunny disposition, joyful and mischievous, and was never heard to complain. He had a sharp sense of humour that he often turned against himself, and he was of a constant and deep kindness.
He leaves a great void for all those who knew him and in particular for the computational biology community in which he was respected and loved.
Our thoughts go to his closest colleagues and particularly to his family.
An image-based high-content screening for compounds targeting Toxoplasma gondii repurposed inhibitors effective against the malaria parasite Plasmodium falciparum
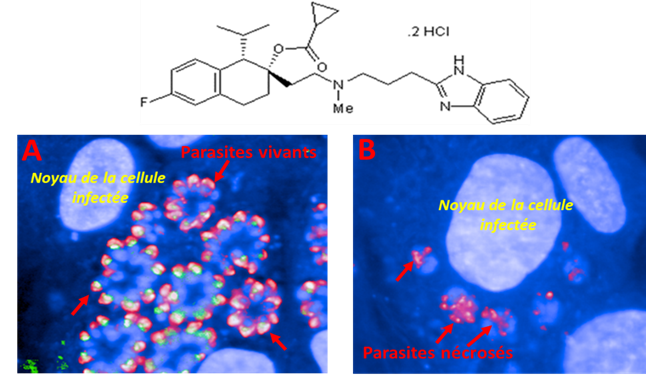
Apicomplexan parasites contain specific secretory organelles called rhoptries and micronemes, whose biogenesis depends on the unique receptor sortilin TgSORT. In the present study, we took advantage of the subcellular polarity of the parasite to engineer a clonal transgenic Toxoplasma line that expresses simultaneously the green fluorescent protein TgSORT-GFP in the post-Golgi-endosome-like compartment and the red fluorescent protein rhoptry ROP1-mCherry near the apical end. We utilized this fluorescent transgenic T. gondii to develop a miniaturized image-based phenotype assay coupled to an automated image analysis. By applying this methodology to 1,120 compounds, we identified 12 that are capable of disrupting the T. gondii morphology and inhibiting intracellular replication. Analysis of the selected compounds confirmed that all 12 are kinase inhibitors and intramembrane pumps, with some exhibiting potent activity against Plasmodium falciparum. Our findings highlight the advantage of comparative and targeted phenotypic analysis involving two related parasite species as a means of identifying molecules with a conserved mode of action.
More information: https://doi.org/10.3389/fcimb.2023.1102551
Contact: Stanislas Tomavo <stanislas.tomavo@i2bc.paris-saclay.fr>
Dual-uptake mode of the antibiotic phazolicin by Gram-negative bacteria prevents resistance acquisition
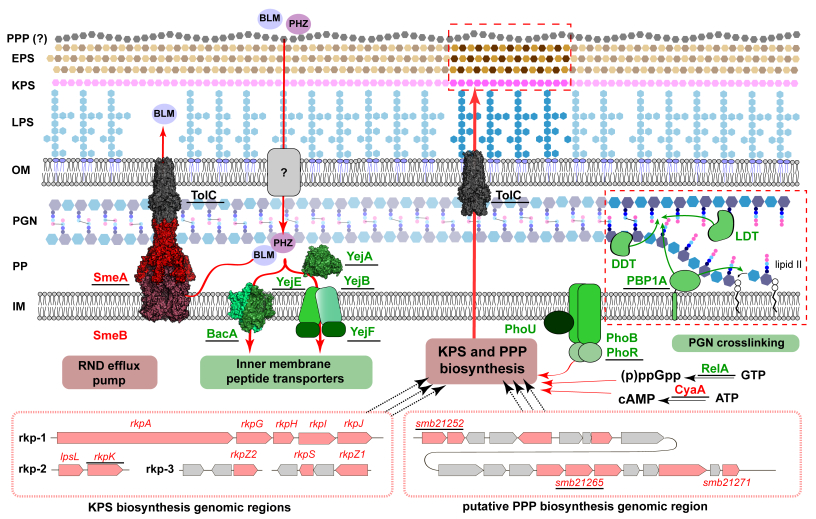
Genetic screens and transposon sequencing have revealed multiple bacterial envelope functions in Sinorhizobium meliloti that contribute to the uptake efficiency of the ribosome-inhibiting antimicrobial peptide phazolicin, explaining why resistance to this molecule is improbable.
Many bacteria produce antimicrobial peptides to eliminate competitors and create an exclusive niche. These peptides act by either membrane disruption or by inhibiting essential intracellular processes. The Achilles heel of the latter type of antimicrobials is their dependence on transporters to enter the susceptible cells because transporter inactivation is sufficient to obtain resistance. Here, we show that a rhizobial ribosome-targeting peptide phazolicin (PHZ) uses two different transporters, BacA and YejABEF, to get into the cells of a symbiotic bacterium Sinorhizobium meliloti. This dual entry mode dramatically reduces the probability of the appearance of PHZ-resistant mutants. Since these transporters are also crucial for S. meliloti symbiotic association with host plants, their inactivation in natural settings is strongly disfavoured, making PHZ an attractive lead for the development of biocontrol agents for agriculture.
More information: https://journals.asm.org/doi/10.1128/mbio.00217-23
Contact: Peter Mergaert <peter.mergaert@i2bc.paris-saclay.fr>
Manganese concentration affects chloroplast structure and the photosynthetic apparatus in Marchantia polymorpha

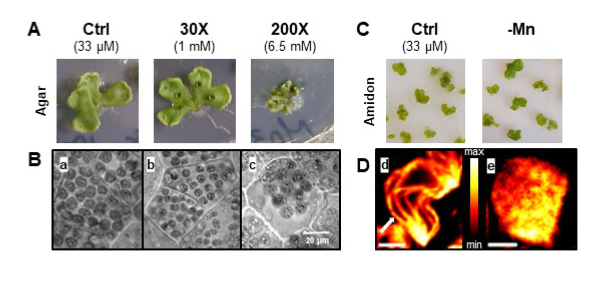
Non-optimal manganese concentration affects photosynthetic electron transport and changes the ultrastructure of the chloroplast as revealed by super-resolution fluorescence microscopy.
Manganese (Mn) is an essential metal for plant growth. It plays a major role in the chloroplast at the level of Photosystem II of the photosynthetic electron transport chain where it forms the Mn4CaO5 cluster, the site of water oxidation. Mn deficiency affects specifically photosynthesis while Mn excess is toxic for the plant. We studied Mn homeostasis in the liverwort Marchantia polymorpha whose simple morphology (absence of cuticle and archaic root system) allows a rapid and uncontrolled absorption. The determination of Mn in thalli and chloroplasts showed a high capacity of uptake (beyond even some metal hyperaccumulating plants) and resistance to stress, in particular thanks to some essential metabolites as shown by GC-MS analyses. In vivo super-resolution fluorescence microscopy (SRM) and transmission electron microscopy showed a change in chloroplast size and a disorganization of the thylakoid membranes. Mn deficiency increased the ratio of photosystem I compared to photosystem II. This alteration seems to favour cyclic electron flow around photosystem I and helps to protect the photosynthetic electron transport chain against photodamage. In conclusion, Marchantia polymorpha is an interesting model plant to study metal homeostasis and to be used by new analytical methods (MRS). In this organism, Mn plays an important role in thylakoid organization and photosynthetic electron transport.
More information: doi: 10.1093/plphys/kiad052
Contact: Anja KRIEGER-LISZKAY <anja.liszkay@i2bc.paris-saclay.fr>
Recipient chromosome-encoded UvrD helicase plays a role in plasmid acquisition by conjugationa


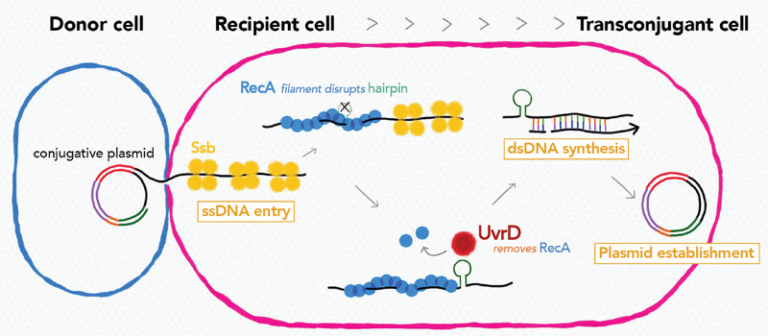
Research on conjugation unveiled that recipient chromosome-encoded UvrD helicase plays role in plasmid acquisition, shedding light on important DNA actions in the early stage of transconjugant cell establishment.
Conjugative plasmids are a major dissemination source of resistance to antibiotics in bacterial populations. Even though they encode genes necessary and sufficient for their horizontal transmission, other environmental and genetic factors can alter the efficiency of plasmid transmission. In this study by using Transposon-insertion site sequencing and genetics experiments, we showed that defects in a conserved DNA helicase, UvrD impairs transfer of various conjugative plasmids. This was observed specifically in the recipient cell, not in the donor cell. During conjugation, one strand of plasmid DNA transfers from the donor to the recipient cell. By time-lapse microscopy with sophisticated monitoring tools to detect single-stranded (ss) and double-stranded (ds)DNA, we showed that UvrD plays a role in successful ss to ds conversion of the acquired DNA. An interesting spin is that UvrD homologs are well-known to be multifunctional and many of them are involved in different DNA repair pathways. Our work showed that in addition to its role in maintaining genome integrity, UvrD is also key for the establishment of horizontally acquired DNA that drives genome diversity and evolution.
More information: https://doi.org/10.1093/nar/gkad075
Contact: Yoshi Yamaichi <yoshiharu.yamaichi@i2bc.paris-saclay.fr>
A paralog of Pcc1 is the fifth core subunit of the KEOPS tRNA modifying complex in Archaea
Discovery of the fifth subunit of the archaeal KEOPS complex involved in the synthesis of a key tRNA modification in Archaea.
In Archaea and Eukaryotes, the synthesis of a universal tRNA modification, N6-threonyl-carbamoyl adenosine (t6A), is catalyzed by the KEOPS complex composed of Kae1, Bud32, Cgi121 and Pcc1. A fifth subunit, Gon7, is found only in Fungi and Metazoa. Here, we identify and characterize a fifth KEOPS subunit in Archaea. This protein, dubbed Pcc2, is a paralog of Pcc1 and is widely conserved in Archaea. Pcc1 and Pcc2 form a heterodimer in solution, and show modest sequence conservation but very high structural similarity. The five-subunit archaeal KEOPS does not form dimers but retains robust tRNA binding and t6A synthetic activity. Pcc2 can substitute for Pcc1 but the resulting KEOPS complex is inactive, suggesting a distinct function for the two paralogs. Comparative sequence and structure analyses point to a possible evolutionary link between archaeal Pcc2 and eukaryotic Gon7. Our work indicates that Pcc2 regulates the oligomeric state of the KEOPS complex, a feature that seems to be conserved from Archaea to Eukaryotes.
More information: https://rdcu.be/c4BFl
Contact: Tamara BASTA-LE BERRE <tamara.basta@i2bc.paris-saclay.fr>
Rabies virus P protein binds to TBK1 kinase
and interferes with the formation of innate immunity-related liquid condensates
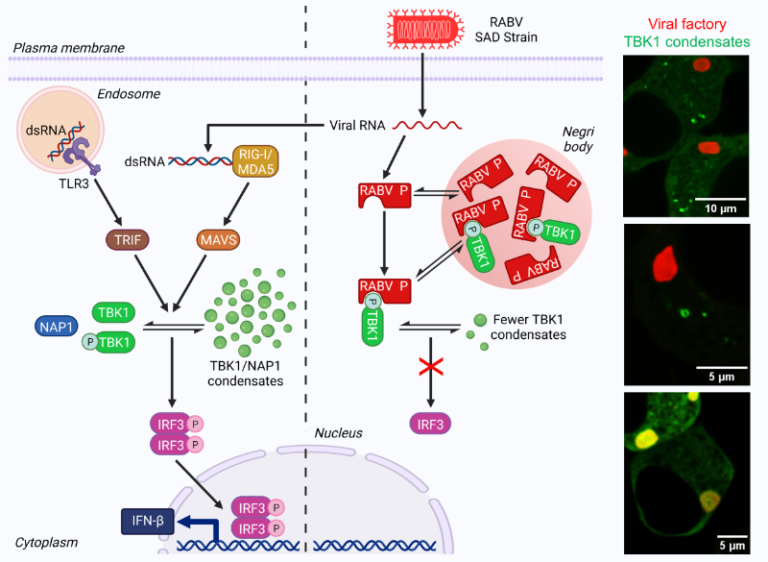
Rabies virus P protein binds TBK1, the IRF3 phosphorylating kinase, and thus inhibits interferon induction. In doing so, P interferes with the formation of TBK1-containing liquid condensates, formed upon viral infection or dsRNA detection by innate immunity.
Viruses must overcome the interferon(IFN)-mediated antiviral response to replicate and spread within their host. Rabies virus (RABV) phosphoprotein P is known to inhibit interferon induction by blocking the phosphorylation of IRF3 transcription factor.
We investigated the molecular bases of this inhibition. For this, we took advantage of the distinct phenotype of two RABV strains (SAD and CVS) with respect to their ability to inhibit IFN induction. We demonstrated that P-SAD, but not P-CVS, inhibits IFN induction. A single amino acid substitution in position 179 is responsible for the difference of phenotype observed between the two strains. Therefore, a recombinant SAD-S179P (in which P-SAD is replaced by its S179P inhibitory version) shows a growth defect in IFN-producing cells.
Using protein mass spectrometry, we identified TBK1 as a protein that preferentially interacts with the inhibitory forms of P. TBK1 is the kinase that phosphorylates IRF3 and sits at the crossroads of many interferon induction pathways. Mutations of TBK1 phosphorylation sites abolish P binding.
More importantly, we demonstrate that upon RABV infection or detection of dsRNA by innate immunity sensors, TBK1 and its adapter proteins NAP1 and SINTBAD form dynamic cytoplasmic condensates that have liquid properties. These condensates can form larger aggregates having ring-like structures in which NAP1 and TBK1 exhibit locally restricted movement. P binding to TBK1 interferes with the formation of these structures.
This work demonstrates that proteins of the signaling pathway leading to interferon induction transiently form liquid condensates that can be targeted by viruses.
More information: https://www.cell.com/cell-reports/pdfExtended/S2211-1247(22)01850-2
Contacts: Yves GAUDIN, Cécile LAGAUDRIERE <yves.gaudin@i2bc.paris-saclay.fr> <cecile.lagaudriere@i2bc.paris-saclay.fr>
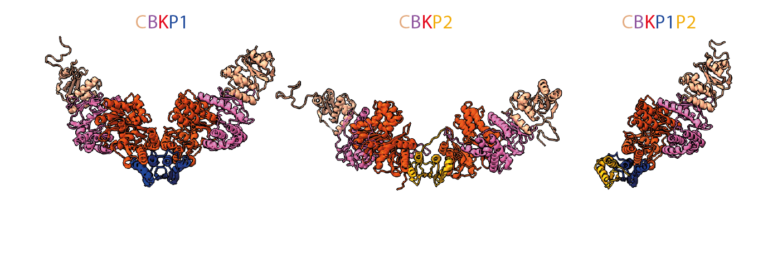
Convergent evolution of bacterial replicative helicase-helicase loader systems
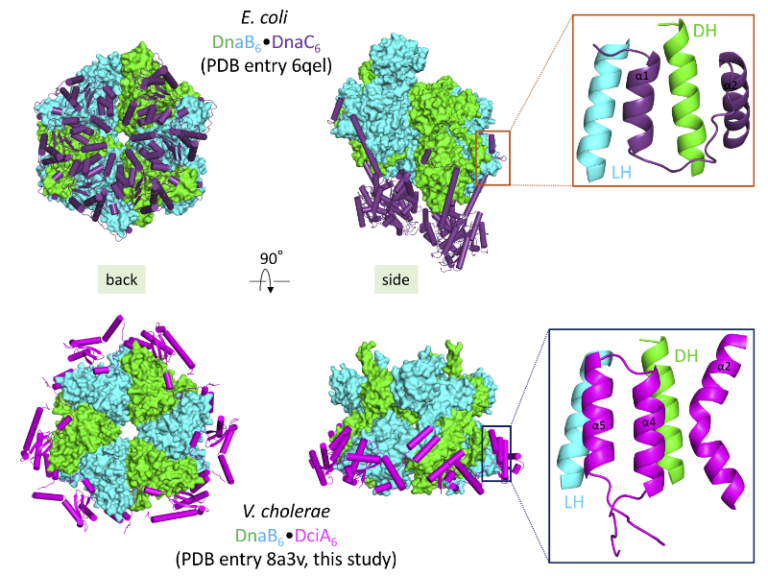
The crystal structure of the DnaB•DciA complex from Vibrio cholerae reveals that the various helicase loaders share the same binding site on bacterial replicative helicases albeit with differences in their loading mechanism.
Ring-shaped DnaB-family helicases play an essential role in bacterial genome replication, by unwinding DNA in front of replication forks. During the initiation step, their loading onto the replication origin depends on dedicated loader proteins. In particular, the well-characterized helicase loader DnaC from Escherichia coli cracks open the DnaB hexameric ring. However, DnaC, which is of phage origin, is an exception in the bacterial world. Instead, most bacteria contain the recently discovered and unrelated helicase loader DciA.
To understand how DciA assists the loading of DnaB onto DNA, the crystal structure of the complex from V. cholerae was determined. Strikingly, DciA binds to the periphery of the DnaB ring, in contrast to other known loaders such as DnaC, which oligomerize and are positioned at the back of the helicase. This discrepancy leads us to consider that the loading mechanism used by DciA, which has still to be elucidated, could differ from those previously described for other loaders. However, it is possible that a third partner, a protein or a nucleic acid, remains to be discovered to fully decipher the function of DciA.
Nevertheless, both DnaC and DciA target the same binding site: the conserved LH-DH module of DnaB helicases. This common binding site for various helicase loaders, as well as the fact that DciA and DnaC can be functionally interchanged in vitro, suggest convergent evolution of the different helicase-helicase loader systems. This could explain how phage loaders were able to efficiently hijack bacterial replicative helicases and replace the ancestral bacterial loader DciA several times during evolution.
More information: https://doi.org/10.1107/S2059798323000281
Contacts: Hélène Walbott, Sophie Quevillon-Cheruel, Stéphanie Marsin <helene.walbott@i2bc.paris-saclay.fr> <sophie.quevillon-cheruel@i2bc.paris-saclay.fr> <stephanie.marsin@i2bc.paris-saclay.fr>
Quelles missions pour la recherche face aux crises de l'anthropocène?
What are the missions for research facing energy-climate change crises ?

Lundi 6 février à 11h dans l’Auditorium du bâtiment 21 – Séminaire en français, ouvert à tous –
Depuis deux siècles, l’utilisation d’une énergie abondante et peu chère permise par les progrès technologiques a progressivement libéré l’homme de sa condition d’agriculteur et conduit à un confort de vie sans équivalent dans l’histoire. Ces changements se sont accompagnés d’une complexification rapide de la société et ont permis notamment l’émergence d’une recherche fonctionnarisée soutenue par des budgets sanctuarisés. Celle-ci, de moins en moins finalisée, nous permet de comprendre le monde qui nous entoure et choisir parmi les futurs possibles. Malheureusement, cette construction sociétale récente vacille car cette énergie abondante, principalement fossile, est en quantité limitée. Par ailleurs, son utilisation déstabilise directement le climat et indirectement nous pousse à dépasser les limites planétaires. Il est évident que le XXIème siècle sera celui de grands changements –subits ou souhaités- car lutter contre le changement climatique veut dire renoncer à une composante majeure de l’abondance actuelle. J’essaierai de montrer que c’est aussi peut être celui de la fin de la recherche telle que nous la pratiquons. Sa forte dépendance à l’énergie la rend particulièrement fragile alors qu’elle est plus que jamais nécessaire pour comprendre l’évolution rapide du monde actuel sous la pression humaine. Mais si, contrainte en moyen et limitée en temps, elle doit disparaître, ne faut-il pas en requestionner les modalités et finalité ? La présentation est largement et librement inspirée des travaux de Jean-Marc Jancovici.
Contact: Sébastien Thomine <sebastien.thomine@i2bc.paris-saclay.fr >
Monday february the 6th in I2BC Auditorium Bld 21- Seminar in french, open to all –
For two centuries, the use of abundant and cheap energy made possible by technological progress has progressively freed man from his condition as a farmer and led to a standard of living without equivalent in history. These changes have been accompanied by a rapid increase in the complexity of society and have led to the emergence of a functional research system supported by dedicated budgets. This research, less and less finalised, allows us to understand the world around us and to choose among possible futures. Unfortunately, this recent societal construction is faltering because this abundant, mainly fossil, energy is in short supply. Moreover, its use directly destabilises the climate and indirectly pushes us to overuse the resources of our planet. It is clear that the 21st century will be one of great changes – whether intentional or unintended – because combating climate change means giving up a major component of the current abundance. I will try to show that it may also be the end of research as we know it. Its heavy dependence on energy makes it particularly fragile, even though it is more necessary than ever to understand the rapid evolution of today’s world under human pressure. Should we reconsider the modalities and purpose of research? The presentation is largely and freely inspired by the work of Jean-Marc Jancovici.
Contact: Sébastien Thomine <sebastien.thomine@i2bc.paris-saclay.fr >
Properties of rabies virus phosphoprotein and nucleoprotein biocondensates formed in vitro and in cellulo
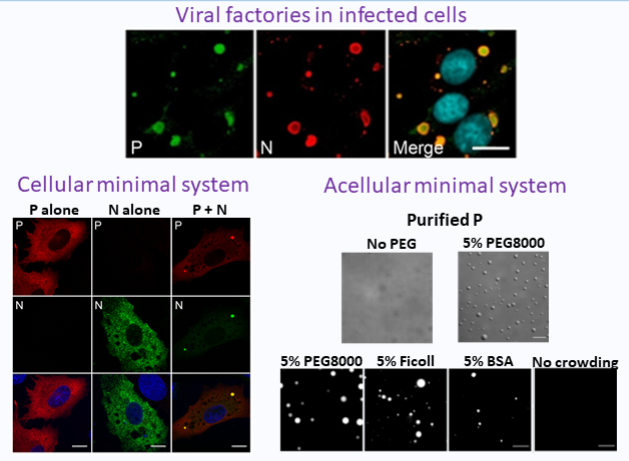
Rabies virus factories are formed by liquid-liquid phase separation. Nevers et al. have developed and characterized two minimal systems, one cellular, the other acellular, which lead to the formation of condensates having the same properties as the viral factories.
Rabies virus (RABV) is a neurotropic virus causing fatal encephalitis in mammals. RABV transcription and replication take place in viral factories, called Negri bodies (NBs), having liquid properties and formed by liquid-liquid phase separation (LLPS). To decipher the molecular bases underlying such processes, it is necessary to design versatile and easy-to-manipulate minimal systems that recapitulate the characteristics of those viral condensates.
Co-expression of RABV nucleoprotein (N, which tightly binds the viral genome) and phosphoprotein (P, a cofactor of the viral polymerase) in cells is sufficient to induce the formation of condensates with properties similar to NBs. This cellular minimal system was previously used to identify P domains essential for condensate formation. Here, we constructed fluorescent versions of N and analyzed their dynamics inside the condensates using FRAP. N behaves differently from P as there is no fluorescence recovery of N after photobleaching. We also identified 3 arginine residues and two loops of N involved in condensate formation.
We also demonstrated that in vitro, in crowded environments, purified P as well as purified N0-P complex (in which N is RNA-free) form liquid condensates. P domains required for LLPS in this acellular system are the same as those required in the cellular minimal system. P condensates associate with liposomes, concentrate RNA, and undergo a liquid-gel transition upon ageing. Conversely, N0-P condensates are disrupted upon incubation with RNA. Taken together, our data emphasize the role of P in NBs formation and reveal some physicochemical features of P and N0-P condensates relevant for explaining NBs properties such as their envelopment by cellular membranes at late stages of infection and nucleocapsids ejections from the viral factories.
These minimal systems will be useful to characterize weak interactions between proteins involved in NBs formation using biophysical techniques.
More information: https://journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1011022
Contact: Yves Gaudin <yves.gaudin@i2bc.paris-saclay.fr >
Accurate atomic model of the hepatitis E virus replication polyprotein
AlphaFold modelling of the multi-domain replication polyprotein of hepatitis E virus fosters insights on how this emerging pathogen generates new RNA viral genomes in infected cells.
Structural biology, the field that produces atomic-level structures of biomacromolecules, has undergone several revolutions in the last ten years. The latest, and possibly the most far-reaching, is the development of artificial intelligence-based prediction of atomic structures of proteins from their sequence. The program AlphaFold has thus demonstrated an amazing capability to produce atomic models of proteins whose accuracy is actually on a par with that of experimental structures derived from X-ray crystallography or high resolution cryo-electron microscopy. Previously this could be achieved only for proteins with very high sequence identity to a protein of known structure. This development is thus particularly important for RNA viruses, whose sequences diverge fast and the proteins of which consequently never have high sequence similarity to other proteins of known structures.
Using AlphaFold, we modelled the hepatitis E virus (HEV) pORF1, the 1700-residue polyprotein that contains the necessary enzymatic activities to synthesise new HEV genomes in infected cells. We describe a five-domain protein with now clear boundaries for domains. The first of these, previously assigned to two distinct methyltransferase and Y domains, actually contains in a single domain both the activities necessary to cap the new viral RNAs (Met part), and interspersed membrane-interacting elements (Y part). We thus term this N-terminal domain MetY. The atomic structure of MetY is clearly homologous to the Chikungunya virus nsP1, despite very low sequence identity except in the methyltransferase active site. We propose a dodecameric assembly for MetY, as has been described experimentally for nsP1. MetY would thus form a pore allowing translocation from the membrane replication compartment to the cytosol coupled to capping of newly synthesised RNAs.
Most RNA viruses with genomes similar to HEV’s, such as hepatitis C virus (HCV), Chikungunya virus, Zika virus, or SARS-CoV-2, cleave their replication polyproteins into mature proteins with virally-encoded proteases. Our atomic model suggests that for HEV structural flexibility rather than proteolytic processing would serve to regulate pORF1 functions. The case of HCV has shown that direct-acting antivirals could cure chronic RNA virus infection, such as occurs in most HEV infections of immunocompromised patients. This work is a step forward in defining the molecular targets for such future HEV antivirals.
More information: https://www.sciencedirect.com/science/article/pii/S0042682222002136
Contact: Sonia Fieulaine <sonia.fieulaine@i2bc.paris-saclay.fr> Thibault Tubiana <thibault.tubiana@i2bc.paris-saclay.fr>
How to tell a gene where and when it should be active?
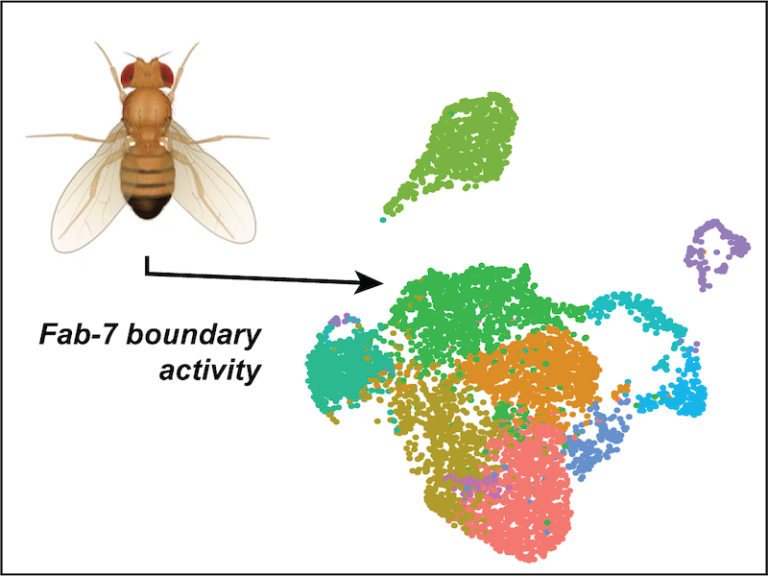
A new study has dissected the complex interactions between regulatory layers that fine-tune the activity of a gene which is essential for the correct formation of the embryo.
The formation of the embryo, from humans to flies, requires a precise orchestration of gene activity: genes need to be turned on at the right time, the right place and at the right level. This regulation of activity incorporates different layers, including the activation of regulatory elements through transcription factor binding, the separation between regulatory regions in the genome through insulator protein binding at boundaries, the epigenetic modifications of large stretches of DNA and the higher-order conformation of chromosomes. How these regulatory layers interact among each other, and particularly how they may instruct each other, remains poorly understood. In this study, PhD student Laura Moniot-Perron in the Chromatin Dynamics group of the I2BC, under the supervision of Daan Noordermeer and Sébastien Bloyer, has studied this question for the Abd-B gene, a prototype gene for embryogenesis. The Abd-B gene encodes a transcription factor that specifies segmental identities along the Antero-Posterior (head-to-tail) body axis. At different positions along this body axis, the gene is activated by different regulatory elements that are located increasingly nearby on the chromosome at more posterior positions. Until now, the question how different regulatory layers are involved in the differential activity along the body axis remained largely unknown. Comparison of cells from different positions along the Antero-Posterior axis revealed the importance of the Fab-7 boundary to create different domains of epigenetic modifications and higher-order chromosome conformation. The resulting cell type-specific organizations differentially activate the different promoters of the Abd-B gene, thereby revealing new possibilities to precisely fine-tune the activity of genes involved in embryogenesis.
More information: https://doi.org/10.1016/j.celrep.2022.111967
Contact: Daan Noordermeer <daan.noordermeer@i2bc.paris-saclay.fr>
New insights into genome annotation in Podospora anserina through re‑exploiting multiple RNA‑seq data

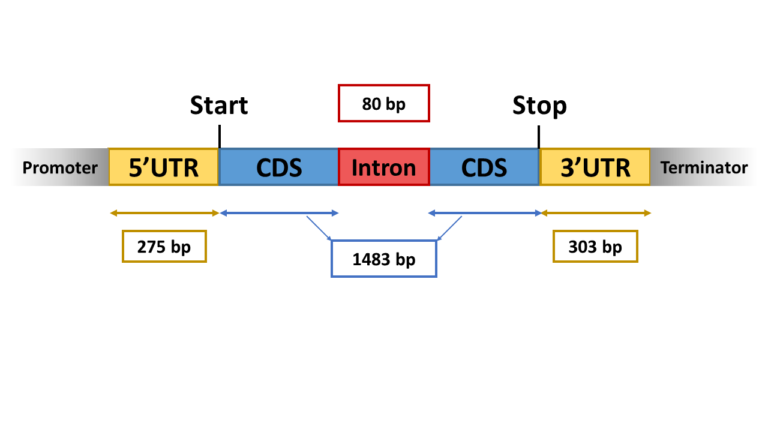
Thanks to multiple RNA-seq data available on public databases, we were able to improve the genome annotation on Podospora anserina and predict important regulatory features such as UTRs, alternative splicing and new transcription units.
Publicly available RNA-seq datasets are often underused although being helpful to improve functional annotation of eukaryotic genomes. This is especially true for filamentous fungi genomes which structure differs from most well annotated yeast genomes. Podospora anserina is a filamentous fungal model, which genome has been sequenced and annotated in 2008. Still, the current annotation lacks information about cis-regulatory elements, including promoters, transcription starting sites and terminators, which are instrumental to integrate epigenomic features into global gene regulation strategies. Here we took advantage of 37 RNA-seq experiments that were obtained in contrasted developmental and physiological conditions, to complete the functional annotation of P. anserina genome. Out of the 10,800 previously annotated genes, 5’UTR and 3’UTR were defined for 7554, among which, 3328 showed differential transcriptional signal starts and/or transcriptional end sites. In addition, alternative splicing events were detected for 2350 genes, mostly due alternative 3’splice sites and 1732 novel transcriptionally active regions (nTARs) in unannotated regions were identified. Our study provides a comprehensive genome-wide functional annotation of P. anserina genome, including chromatin features, cis-acting elements such as UTRs, alternative splicing events and transcription of noncoding regions. These new findings will likely improve our understanding of gene regulation strategies in compact genomes, such as those of filamentous fungi. Characterization of alternative transcripts and nTARs paves the way to the discovery of putative new genes, alternative peptides or regulatory non-coding RNAs.
More information: https://rdcu.be/c2C2M
Contact: Pierre Grognet <pierre.grognet@i2bc.paris-saclay.fr>
Strategic Axes Day

Program (EV)
9h-9h20 : Introduction (amphitheatre, building 21)
9h20-10h20 : Feedback on programs selected in 2021 (amphitheatre, building 21)
9h20-9h50 MEMlessCOM, axis 1 (F. Frottin, Y. Gaudin)
9h50-10h20 EpiRNA, axis 2 (O. Namy)
10h20-10h50 : Coffee break (hall, building 21)
10h50-11h50 : Feedback on programs selected in 2021, continued (amphi, building 21)
10h50-11h20 Macro, axis 1 (C. Le Clainche, J. Ménétrey, AM Tassin, B. Gigant)
11h20-11h50 AssemblyArt, axis 4 (P. Minard, A. Urvoas)
11h50-12h50 : Feedback on programs selected in 2022 (amphi, building 21)
11h50-12h05 ReMembER, axes 1 & 3 (A. Esclatine, F. Giordano)
12h05-12h20 RecoLiPro, axis 1 (S. Bressanelli, G. Lenoir, T. Touzé)
12h20-12h35 AC/DC, axis 2 (S. Bury-Moné, D. Noordermeer)
12h35-12h50 MnAR, axis 4 (A. Krieger-Liszkay)
13h-14h : Lunch in small groups (canteen)
14h-15h : Parallel thematic workshops (amphi and blue room, building 21 + rooms N0-001 et N0-009, building 22)
– Axes/programs, inter-team exchanges and scientific animation
– Axes/programs, technological developments and platforms/facilities
– I2BC axes in the Paris-Saclay environment (links with other institutes, GS, OI, EUGLOH…)
– Scientific themes to be developed (proposals and ideas, within or between axes)
15h-15h30 : Coffee break (hall, building 21)
15h30-17h : : Restitution of the workshops and general discussion (amphitheatre, building 21)
Programme (VF)
9h-9h20 : Introduction (amphi bât 21)
9h20-10h20 : Retours sur les programmes de la vague 2021 (amphi bât 21)
9h20-9h50 MEMlessCOM, axe 1 (F. Frottin, Y. Gaudin)
9h50-10h20 EpiRNA, axe 2 (O. Namy)
10h20-10h50 : Pause-café (hall bât 21)
10h50-11h50 : Retours sur les programmes de la vague 2021, suite (amphi bât 21)
10h50-11h20 Macro, axe 1 (C. Le Clainche, J. Ménétrey, AM Tassin, B. Gigant)
11h20-11h50 AssemblyArt, axe 4 (P. Minard, A. Urvoas)
11h50-12h50 : Retours sur les programmes de la vague 2022 (amphi bât 21)
11h50-12h05 ReMembER, axes 1 & 3 (A. Esclatine, F. Giordano)
12h05-12h20 RecoliPro, axe 1 (S. Bressanelli, G. Lenoir, T. Touzé)
12h20-12h35 AC/DC, axe 2 (S. Bury-Moné, D. Noordermeer)
12h35-12h50 MnAR, axe 4 (A. Krieger-Liszkay)
13h-14h : Déjeuner libre (cantine par petits groupes)
14h-15h : Ateliers parallèles thématiques (amphi et salle bleue bât 21 + salles N0-001 et N0-009 bât 22)
– Axes/programmes, échanges inter-équipes et animation scientifique
– Axes/programmes, développements technologiques et plateformes
– Axes I2BC dans l’environnement Paris-Saclay (liens avec autres instituts, GS, OI, EUGLOH…)
– Thématiques scientifiques à développer (propositions et idées, dans un axe ou inter-axes)
15h-15h30 : Pause-café (hall bât 21)
15h30-17h : Restitution des ateliers et discussion générale (amphi bât 21)
