Summary
- The structure of a Tau fragment bound to tubulin prompts new hypotheses on Tau mechanism and oligomerization
- Ribosome composition plays a key role in EMT
- DciA, the Bacterial Replicative Helicase Loader, promotes LLPS in the presence of ssDNA
- Dual RNA-seq study of the dynamics of coding and non-coding RNA expression during Clostridioides difficile infection in a mouse model
- The ribosome profiling landscape of yeast reveals a high diversity in pervasive translation
- The next-generation sequencing—chess problem
- Perylene-derivative singlet exciton fission in water solution
- Bioorthogonal monomycolate of trehalose disclosed the O-mycoloylation of mycoloyltransferases and other cell envelope proteins in C. glutamicum
- Talin and vinculin combine their activities totrigger actin assembly
- DciA secures bidirectional replication initiation in Vibrio cholerae
- The acetyltransferase SCO0988 controls positively specialized metabolism and morphological differentiation in the model strains Streptomyces coelicolor and Streptomyces lividans.
- De Novo Emerged Gene Search in Eukaryotes with DENSE
- A complex and dynamic redox network regulates oxygen reduction at photosystem I in Arabidopsis
- Impact of the Deletion of Genes of the Nitrogen Metabolism on Triacylglycerol, Cardiolipin and Actinorhodin Biosynthesis in Streptomyces coelicolor
- Molecular insights into the activation of Mre11-Rad50 endonuclease activity by Sae2/CtIP
- Sulfur starvation-induced autophagy in Saccharomyces cerevisiae involvesSAM-dependent signaling and transcription activator Met4
- Prophage induction can facilitate the in vitro dispersal of multicellular Streptomyces structure
- TLN468 changes the pattern of tRNA used to read through premature termination codons in CFTR
- A tumor suppressor that generates subnucleosomes
- BRCA2 stabilizes DMC1 nucleoprotein filaments in meiosis
- SYNTERUPTOR: mining genomic islands for non-classical specialized metabolite gene clusters
- Acinetobacter baumannii satellite phage Aci01-2-Phanie depends on a helper myophage for its multiplication
- Gradual ER calcium depletion induces a progressive and reversible UPR signaling
- Timing is success! Bacteriophage tail completion proteins are essential regulators of viral DNA delivery to host bacteria
- Mechanism of DNA unwinding by MCM8-9 in complex with HROB
- Nuclear receptor signaling via NHR-49/MDT-15 regulates stress resilience and proteostasis in response to reproductive and metabolic cues
- Staphylococcus aureus in the battle for iron: three post-transcriptional regulations at work
- Hundreds of antimicrobial peptides create a selective barrier for insect gut symbionts
- Imprinting gene regulation: Importance of a long non-coding RNA and of DNA methylation levels at an essential CTCF binding site
- Uncoupling programmed DNA cleavage and repair scrambles the Paramecium somatic genome
- Transcription-induced DNA supercoiling clears RNA polymerase’s path in bacterial chromatin
- A Fatty Acid Anabolic Pathway in Specialized-Cells Sustains a Remote Signal that Controls Egg Activation in Drosophila
- A highly conserved ligand-binding site for AccA transporters of antibiotic and quorum-sensing regulator in Agrobacterium leads to a different specificity
- Epigenetics: combining flexible and rigid regions into a single structure to ensure genome replication and stability
- A genome-wide comprehensive analysis of nucleosome positioning in yeast
- Activity of MukBEF for chromosome management in E. coli and its inhibition by MatP
- Cooperation between two modes for DNA replication initiation in the archaeon Thermococcus barophilus
- JAK-STAT-dependent contact between follicle cells and the oocyte controls Drosophila anterior-posterior polarity and germline development
- Transposon sequencing reveals genes enabling insect gut colonization by the symbiont Caballeronia insecticola
- Protein-protein interactions: how to push forward the limits of the revolutionary AlphaFold2 programme?
- The stringent response is strongly activated in the high antibiotic producer, Streptomyces coelicolor.
- Composition of Poxvirus Core Revealed
- Schizosaccharomyces pombe as a fundamental model for research on mitochondrial gene expression: Progress, achievements and outlooks
- Antigen self-anchoring onto bacteriophage T5 capsid-like particles for vaccine design
- Toxoplasma membrane inositol phospholipid binding protein TgREMIND is essential for secretory organelle function and host infection
The structure of a Tau fragment bound to tubulin prompts new hypotheses on Tau mechanism and oligomerization
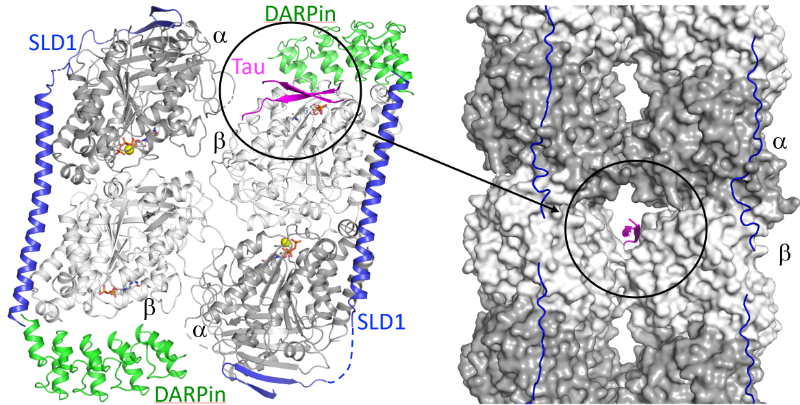
We have identified a novel tubulin surface targeted by the Tau protein. From these results, we formulate new hypotheses on the regulation of microtubules by Tau and on Tau oligomerization.
Tau is a protein involved in the regulation of axonal microtubules in neurons. In pathological conditions, it forms filamentous aggregates which are molecular markers of the Alzheimer’s disease and of related neurodegenerative disorders collectively known as tauopathies. Structures of Tau in fibrils or bound to the microtubule have been reported. We have determined the structure of a Tau construct comprising the PHF6 motif, an hexapeptide involved in Tau aggregation, as a complex with tubulin. This Tau fragment binds as a dimer to a new site which, when transposed to the microtubule, would correspond to a pore between protofilaments. These results raise new hypotheses on Tau-induced microtubule assembly and stabilization and on Tau oligomerization. They reconcile apparently contradictory data from the literature, in particular concerning the coexistence of different binding modes of Tau to the microtubule.
More information: https://academic.oup.com/pnasnexus/article/3/11/pgae487/7850850
Contact: Benoît GIGANT <benoit.gigant@i2bc.paris-saclay.fr>
Ribosome composition plays a key role in EMT

Ribosome remodeling drives cancer cell transformation: A single protein, RPL36A, triggers EMT.
Epithelial-mesenchymal transition (EMT) is a biological process whereby a cell loses its usual characteristics to acquire properties allowing it to move. EMT is involved in embryo formation and tissue repair in adults. However, when altered, EMT contributes to the formation of fibrosis or, in the case of a tumor cell, to the formation of metastases. In cancers, this change is comparable to a cellular metamorphosis that allows cancer cells to become more aggressive and resistant to treatments. EMT is therefore a key player in tumor progression.
EMT is extensively studied at the molecular and cellular levels because a better understanding of this process will enable the development of new therapeutic approaches. However, the translational changes that occur during EMT have so far been very poorly studied.
In this work, we have identified that the overexpression of a single ribosomal protein, called RPL36A, is sufficient to induce EMT. We have also highlighted the various translational changes that occur during EMT.
Our work reveals the importance of ribosome remodeling itself in finely modulating translation during the change in cellular identity that occurs during EMT.
More information: https://www.pnas.org/doi/10.1073/pnas.2408114121
Contact: Olivier NAMY <olivier.namy@i2bc.paris-saclay.fr>
DciA, the Bacterial Replicative Helicase Loader, promotes LLPS in the presence of ssDNA

This study shows that DciA, the bacterial replicative helicase loader, promotes condensates in the presence of DNA. This opens the way to the possibility of non-membrane compartments in bacteria. The goal could be to concentrate the players involved in replication and thus facilitate it.
The loading of the bacterial replicative helicase DnaB is an essential step for genome replication and depends on the assistance of accessory proteins. Several of these proteins have been identified across the bacterial phyla. DciA is the most common loading protein in bacteria, yet the one whose mechanism is the least understood. We have previously shown that DciA from Vibrio cholerae is composed of a globular domain followed by an unfolded extension and demonstrated its strong affinity for DNA. Here, drawing on the skills of two I2BC facilities, Light microscopy (Imagerie-Gif) and PIM (Structural biology), we characterize the condensates formed by VcDciA upon interaction with a short single-stranded DNA substrate. We demonstrate the fluidity of these condensates using light microscopy and address their network organization through electron microscopy, thereby bridging events to conclude on a liquid-liquid phase separation behavior. Additionally, we observe the recruitment of DnaB in the droplets, concomitant with the release of DciA. We show that the well-known helicase loader DnaC from Escherichia coli is also competent to form these phase-separated condensates in the presence of ssDNA. Our phenomenological data are still preliminary as regards the existence of these condensates in vivo, but open the way for exploring the potential involvement of DciA in the formation of non-membrane compartments within the bacterium to facilitate the assembly of replication players on chromosomal DNA.
More information: https://pubmed.ncbi.nlm.nih.gov/39603490/
Contact: Sophie CHERUEL & Stéphanie MARSIN <sophie.quevillon-cheruel@i2bc.paris-saclay.fr> & <stephanie.marsin@i2bc.paris-saclay.fr>
Dual RNA-seq study of the dynamics of coding and non-coding RNA expression during Clostridioides difficile infection in a mouse model

First adaptation of in vivo dual transcriptomics to human enteropathogen C. difficile revealed dynamics of gene expression in pathogen, its host, and gut microbiota composition and identified the novel RNA components shaping C. difficile virulence and host response.
Clostridioides difficile is a major cause of nosocomial infections associated with antibiotic therapy classified as an urgent antibiotic resistance threat. This pathogen interacts with host and gut microbial communities during infection, but the mechanisms of these interactions remain largely to be uncovered. Noncoding RNAs contribute to bacterial virulence and host responses, but their expression has not been explored during C. difficile infection. This collaborative work of I2BC and Micalis laboratories took advantage of the conventional mouse model of C. difficile infection to look simultaneously to the dynamics of gene expression in pathogen, its host, and gut microbiota composition, providing valuable resources for future studies. We identified a number of ncRNAs that could mediate the adaptation of C. difficile inside the host and the crosstalk with the host immune response. Promising inflammation markers and potential therapeutic targets emerged from this work open new directions for RNA-based and microbiota-modulatory strategies to improve the efficiency of C. difficile infection treatments.
More information: https://journals.asm.org/doi/10.1128/msystems.00863-24
Contact: Olga SOUTOURINA <olga.soutourina@i2bc.paris-saclay.fr>
The ribosome profiling landscape of yeast reveals a high diversity in pervasive translation
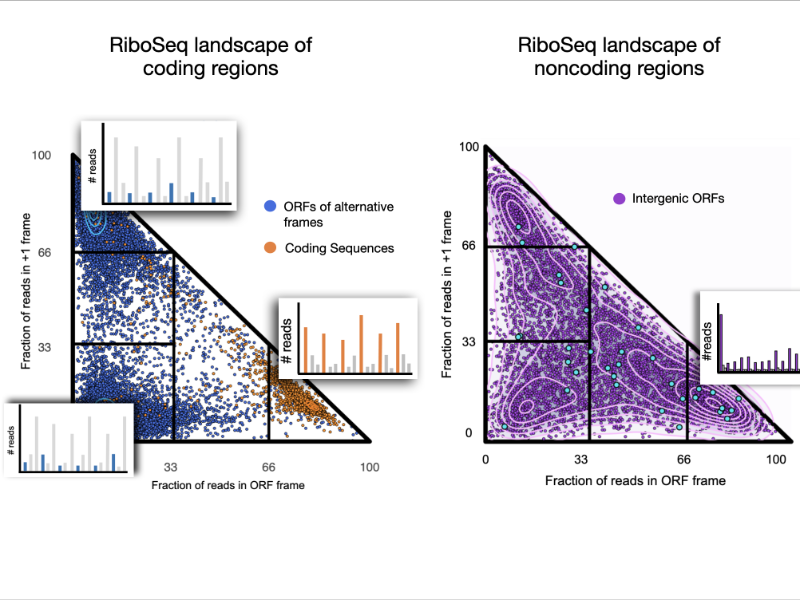
We explored the ribosome profiling landscape of yeast and showed that noncoding regions are associated with a wide diversity of translation signals among which some led to detectable protein products.
Pervasive translation is a widespread phenomenon that plays a critical role in the emergence of novel microproteins, but the diversity of translation patterns contributing to their generation remains unclear. Based on 54 ribosome profiling (Ribo-Seq) datasets, we investigated the yeast Ribo-Seq landscape using a representation framework that allows the comprehensive inventory and classification of the entire diversity of Ribo-Seq signals, including non-canonical ones. We show that if coding regions occupy specific areas of the Ribo-Seq landscape, noncoding regions encompass a wide diversity of Ribo-Seq signals and, conversely, populate the entire landscape. Our results show that pervasive translation can, nevertheless, be associated with high specificity, with 1055 noncoding ORFs exhibiting canonical Ribo-Seq signals. Using mass spectrometry under standard conditions or proteasome inhibition with an in-house analysis protocol, we report 239 microproteins originating from noncoding ORFs that display canonical but also non-canonical Ribo-Seq signals. Each condition yields dozens of additional microprotein candidates with comparable translation properties, suggesting a larger population of volatile microproteins that are challenging to detect. Our findings suggest that non-canonical translation signals may harbor valuable information and underscore the significance of considering them in proteogenomic studies. Finally, we show that the translation outcome of a noncoding ORF is primarily determined by the initiating codon and the codon distribution in its two alternative frames, rather than features indicative of functionality. Our results enable us to propose a topology of a species’ Ribo-Seq landscape, opening the way to comparative analyses of this translation landscape under different conditions.
More information: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03403-7
Contact: Anne LOPES <anne.lopes@i2bc.paris-saclay.fr>
The next-generation sequencing—chess problem

By comparing the collection of Next-Generation Sequencing data to the superimposition of several chess games, we demonstrate the limits of current temporal analysis approaches.
The development of next-generation sequencing (NGS) technologies paved the way for studying the spatiotemporal coordination of cellular processes along the genome. However, data sets are commonly limited to a few time points, and missing information needs to be interpolated. Most models assume that the studied dynamics are similar between individual cells, so that a homogeneous cell culture can be represented by a population-wide average. Here, we demonstrate that this understanding can be inappropriate. We developed a thought experiment—which we call the NGS chess problem—in which we compare the temporal sequencing data analysis to observing a superimposed picture of many independent games of chess at a time. The analysis of the spatiotemporal kinetics advocates for a new methodology that considers DNA-particle interactions in each cell independently even for a homogeneous cell population.
More information: https://academic.oup.com/nargab/article/6/4/lqae144/7833696
Contact: Julie SOUTOURINA <julie.soutourina@i2bc.paris-saclay.fr>
Perylene-derivative singlet exciton fission in water solution

Ultrafast formation of triplet statates by singlet fission in water solution.
We provide evidence that singlet fission—a process where one excited state splits into two—can occur in water-soluble compounds. Specifically, we studied perylene-3,4,9,10-tetracarboxylate, a molecule that forms transient disordered dimers. Using a combination of advanced techniques like time-resolved absorption and fluorescence spectroscopy, nuclear magnetic resonance (NMR) spectroscopy, and theoretical modeling, we were able to detect the key features of singlet fission and map out the behavior of the molecular assemblies. Our results show that the constant structural changes within these dimers play a crucial role in steering the process toward either singlet fission or charge separation. The quantum efficiency of producing triplet states is over 100%, with these states lasting for nanoseconds—highlighting how disordered systems can drive highly efficient photophysical processes.
More information: https://pubs.rsc.org/en/content/articlehtml/2024/sc/d4sc04732j
Contact: Manuel LLANSOLA <manuel.llansola@I2bc.paris-saclay.fr>
Bioorthogonal monomycolate of trehalose disclosed the O-mycoloylation of mycoloyltransferases and other cell envelope proteins in C. glutamicum

Discovering the mycoloylome of Corynebacterium glutamicum : a new type of lipidated-modified proteins.
Protein mycoloylation is a lipidation pathway exclusively observed in Mycobacteriales, an order of bacteria that includes several human pathogens. These bacteria possess a distinctive outer membrane, known as the mycomembrane, composed of very long-chain fatty acids called mycolic acids. It has been demonstrated that a few mycomembrane proteins undergo covalent modification with mycolic acids in the model organism Corynebacterium glutamicum, through the action of mycoloyltransferase MytC. This PTM represents the first example of protein O-acylation in prokaryotes and also the first example of protein modification by mycolic acid. Here, we have developed a unique bioorthogonal mycolate donor featuring the natural mycolic acid pattern, enabling direct unambiguous transfer of the lipid moiety to its acceptors and efficient metabolic labeling and enrichment of MytC protein substrates. Mass spectrometry analysis of the labeled proteins and comparative proteomic analysis of the cell envelope proteome between wild-type and ∆mytC strains identified an unbiased list of 21 proteins likely mycoloylated in the cell. The robustness of our approach is demonstrated by the successful biological validation of mycoloylation in 6 random candidate proteins within wild-type cells, revealing the characteristic profile of proteins modified with natural mycolates. These findings provide interesting insights into the significance of this new lipidation pathway and pave the way for understanding their function, especially concerning the mycoloyltransferase family that includes the essential Antigen85 enzymes in Mycobacteria.
More information: https://doi.org/10.1021/acschembio.4c00502.
Contact: Cécile LABARRE <cecile.labarre@i2bc.paris-saclay.fr>
Talin and vinculin combine their activities totrigger actin assembly

In cells, focal adhesions (FAs) strengthen their connection with the actin cytoskeleton to resist force. By combining biochemistry and cell biology, the authors show that the proteins talin and vinculin control actin assembly, thus reinforcing the anchoring of actin to FAs.
Focal adhesions (FAs) strengthen their link with the actin cytoskeleton to resistforce. Talin-vinculin association could reinforce actin anchoring to FAs bycontrolling actin polymerization. However, the actin polymerization activity ofthe talin-vinculin complex is not known because it requires the reconstitutionof the mechanical and biochemical activation steps that control the associa-tion of talin and vinculin. By combining kinetic and binding assays with singleactinfilament observations in TIRF microscopy, we show that the associationof talin and vinculin mutants, mimicking mechanically stretched talin andactivated vinculin, triggers a sequential mechanism in whichfilaments arenucleated, capped and released to elongate. In agreement with these obser-vations, FRAP experiments in cells co-expressing the same constitutivemutants of talin and vinculin revealed accelerated growth of stressfibers. Ourfindings suggest a versatile mechanism for the regulation of actin assembly inFAs subjected to various combinations of biochemical and mechanical cues.
More information: https://www.nature.com/articles/s41467-024-53859-1
Contact: Christophe LE CLAINCHE <christophe.leclainche@i2bc.paris-saclay.fr>
DciA secures bidirectional replication initiation in Vibrio cholerae

DciA secures bidirectional replication initiation in Vibrio cholerae.
Replication is initiated bidirectionally in the three domains of life by the assembly of two replication forks at an origin of replication. This is made possible by the recruitment of two replicative helicases to a nucleoprotein platform built at the origin of replication with the initiator protein. The reason why replication is initiated bidirectionally has never been experimentally addressed due to the lack of a suitable biological system. Using genetic and genomic approaches, we show that upon depletion of DciA, replication is no longer initiated bidirectionally at the origin of replication of Vibrio cholerae chromosome 1. We show that following unidirectional replication on the left replichore, nascent DNA strands at ori1 anneal to each other to form a double-stranded DNA end. While this DNA end can be efficiently resected in recB+ cells, only a few cells use it to trigger replication on the right replichore. In most DciA-depleted cells, chromosome 1 is degraded leading to cell death. Our results suggest that DciA is essential to ensuring bidirectional initiation of replication in bacteria, preventing a cascade of deleterious events following unidirectional replication initiation.
More information: https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkae795/7759985
Contact: Amelie BESOMBES amelie.besombes@i2bc.paris-saclay.fr
The acetyltransferase SCO0988 controls positively specialized metabolism and morphological differentiation in the model strains Streptomyces coelicolor and Streptomyces lividans.

Acetylation of some proteins plays a positive role in the regulation of the production of specialized metabolites and in the morphological differentiation of Streptomyces.
Streptomyces species possess in their genomes numerous silent biosynthetic pathways likely to direct the biosynthesis of novel bioactive specialized metabolites. Enhancing the expression of these cryptic pathways might lead to the discovery of most needed novel antibiotics to limit the spreading of pathogenic bacteria resistant to most antibiotics in current use.
We characterized, an acetyl transferase (SCO0988) whose over-expression led to enhanced antibiotic production and accelerated sporulation of a low antibiotic producing strain, Streptomyces lividans, while the deletion of this gene had opposite effects in a strong antibiotic producer, Streptomyces coelicolor. Comparative analysis of the acetylome of the S. lividans strain overexpressing SCO0988 with that of the original strain revealed that 92 peptides, belonging to various proteins involved in different cellular processes, were exclusively acetylated in the strain over-expressing SCO0988. This led to the definition of a consensus SCO0988 acetylation site (v/q-P-l/g-D-p/e-KR-v/l-h/f-y/a-t/m). Among the acetylated proteins, BldKB was found to be over-acetylated on four lysine residues including Lys425 in the strain overexpressing SCO0988. BldKB is the solute binding protein of an ABC transporter involved in the up-take of an oligopeptide that plays a role in the triggering of a quorum sensing regulatory cascade controlling positively specialized metabolism, aerial mycelium formation and sporulation in S. coelicolor. A bldKB mutant has a reduced antibiotic production and does not develop aerial mycelium. The bldKB mutant phenotypes can be complemented by the native bldKB gene but not by a variant of this gene encoding a protein in which Lys425 was replaced by Arg (that cannot be acetylated) or by Glu (a putative structural analogue of acetylated lysine). Our study thus demonstrated that Lys425 is critical for BldKB function but could not clearly establish the impact of Lys425 acetylation on BldKB function.
More information: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11303876/
Contact: Marie-Joëlle VIROLLE marie-joelle.virolle@i2bc.paris-saclay.fr
De Novo Emerged Gene Search in Eukaryotes with DENSE

DENSE with your favorite genome and uncover its de novo genes! Try our fully automated tool for DE Novo gene SEarch and discover useful metrics to help design your search!
The recent discovery of de novo emerged genes, which originate from previously noncoding regions of DNA, challenges traditional views of species evolution. Indeed, the hypothesis of neutrally evolving sequences giving rise to functional proteins is highly unlikely. However, the accumulation of sequencing data revealed that they are widespread, being detected in various eukaryotic species and numerous, with several dozens of examples reported for different organisms. This conundrum has sparked numerous studies to quantify and characterize these genes, aiming to understand their functional roles and contributions to genome evolution. Yet, no fully automated pipeline for their identification is available. Therefore, we introduce DENSE (DE Novo emerged gene SEarch), an automated Nextflow pipeline based on two distinct steps: detection of taxonomically restricted genes (TRGs) through phylostratigraphy, and filtering of TRGs for de novo emerged genes via genome comparisons and synteny search. DENSE is available as a user-friendly command-line tool, while the second step is accessible through a web server upon providing a list of TRGs. Highly flexible, DENSE provides various strategy and parameter combinations, enabling users to adapt to specific configurations or define their own strategy through a rational framework, facilitating protocol communication, and study interoperability. We apply DENSE to seven model organisms, exploring the impact of its strategies and parameters on de novo gene predictions. This thorough analysis across species with different evolutionary rates reveals useful metrics for users to define input datasets, identify favorable/unfavorable conditions for de novo gene detection, and control potential biases in genome annotations. Additionally, predictions made for the seven model organisms are compiled into a requestable database, which we hope will serve as a reference for de novo emerged gene lists generated with specific criteria combinations.
More information: https://academic.oup.com/gbe/article/16/8/evae159/7745841
Contact: Anne LOPES anne.lopes@i2bc.paris-saclay.fr
A complex and dynamic redox network regulates oxygen reduction at photosystem I in Arabidopsis

A system of redox enzymes facilitates a series of reactions that regulate oxygen reduction at photosystem I in plants.
In a study published in Plant Physiology, scientists from I2BC and IPS2 studied the redox regulation of superoxide production at the photosynthetic electron transfer level (pseudocyclic flow). Plants must optimise their molecular strategies to adapt to the light conditions of their environment. Reactive oxygen species (ROS) can play a role as signalling molecules but can also be potentially harmful. To manage light-induced ROS and maintain proper photosynthetic function, thiol-dependent redox enzymes play a crucial role in the redox regulation of the chloroplast. It has been shown that the main redox enzymes involved in controlling the ROS generated during photosynthesis are m-type thioredoxins, NADPH-dependent reductase C (NTRC) and peroxiredoxin 2-Cys. The interaction of these enzymes functions as a redox regulatory network, where 2-Cys PRX can be reduced by NTRC and Trx but can also re-oxidise the reduced thioredoxins, thus providing a system for rapid acclimatisation to changes in the light regime. The researchers showed that the membrane localisation of Trx m and NTRC varies as a function of photoperiod. In addition, their results show that it is the PSI itself that is redox regulated. The new results of this study make it possible to generate a new model of PSI redox regulation. This model may guide further research into the redox regulation of alternative electron transport pathways under conditions of fluctuating light or abiotic stress.
More information: DOI: 10.1093/plphys/kiae501
Contact: Anja KRIEGER-LISZKAY anja.liszkay@i2bc.paris-saclay.fr
Impact of the Deletion of Genes of the Nitrogen Metabolism on Triacylglycerol, Cardiolipin and Actinorhodin Biosynthesis in Streptomyces coelicolor.

Analysis of the consequences of the deletion of genes involved in nitrogen metabolism on the content of phospholipids or of storage lipids of Streptomyces coelicolor illustrates the complex links exiting between carbon, nitrogen and phosphate metabolisms.
Since nitrogen (N) limitation is known to be an important trigger of storage lipid / triacylglycerol (TAG) accumulation, in most microorganisms, we assessed the global lipid content of 21 strains derived from Streptomyces coelicolor deleted for genes involved in N metabolism. These strains were grown in the classical R2YE medium that is N and phosphate (P) limited. Seven of these strains had a higher TAG content than the original strain. These strains were either deleted for genes encoding proteins involved 1) in polyamine degradation (GlnA2/SCO2241, GlnA3/SCO6962, GlnA4/SCO1613); 2) in protein degradation (Pup/SCO1646); 3) in the regulation of nitrogen metabolism (GlnE/SCO2234 and GlnK/SCO5584) or 4) encoding the global regulator DasR/SCO5231 that controls negatively the degradation of N-acetylglucosamine, a constituent of peptidoglycan. Five of these strains, with the exception of the glnA2 and pup mutants, had a lower cardiolipin (CL) content than the original strain. This suggested that in these strains the biosynthesis of TAG, molecule devoided of (P) groups is prevalent over that of CL that bears 2 (P) groups, thus allowing higher (P) availability. The deletion of pup, glnA2, glnA3, and glnA4 was correlated, in Pi limitation, with an increase in the total production of the blue polyketide actinorhodin (ACT), whereas ACT production was strongly reduced in the glnA3 mutant in (P) proficiency and was totally abolished in the dasR mutant in both (P) conditions. Altogether, our data suggest that N limitation linked to the deletion of genes mentioned above results into high oxidative stress that triggers storage of acetylCoA as TAG to reduce the activity of the oxidative metabolism, generator of oxidative stress, as well as the biosynthesis of ACT that has anti-oxidant properties. At last, the reduced ACT biosynthesis of the glnA3 mutant, is thought to be due to its high spermine and spermidine content, molecules known to protect the cell against oxidative stress.
More information: doi.org/10.3390/microorganisms12081560
Contact: Marie-Joëlle VIROLLE marie-joelle.virolle@i2bc.paris-saclay.fr
Molecular insights into the activation of Mre11-Rad50 endonuclease activity by Sae2/CtIP

Early Steps of DNA Recombination: AlphaFold2 breaks a lock.
The human MRN nuclease, formed by the association of two proteins, Mre11 and Rad50, is a key player in homologous recombination and meiosis, two processes that utilize DNA break repair mechanisms. In eukaryotes, the Mre11 nuclease is equipped with a molecular lock that controls the activation of the enzyme: its activation is triggered during certain phases of the cell cycle (entry into G2, when DNA is duplicated to allow for the recombination process), when the lock is phosphorylated. This locking protein is called CtIP in humans and Sae2 in the yeast Saccharomyces cerevisiae. It is a protein with a largely disordered structure, which remained a significant challenge for structural characterization for a number of years.
In a study published in Molecular Cell, teams from I2BC, the Curie Institute, and IRB shed light on how the human MRN nuclease functions. The I2BC team modeled the structure of the yeast MRN complex using the AlphaFold2 algorithm. Interestingly, the program hesitates when modeling between two states: inactive (auto-inhibited) and active. Notably, the addition of phosphorylated Sae2 favors the active conformation in AlphaFold2’s predictions: Sae2 appears to unlock the MRN complex by establishing a synergistic set of interactions centered on the phosphorylation of a serine residue in Sae2. The structural predictions obtained with AlphaFold2, supported by in vitro and in vivo experiments, highlight that the phosphorylated Sae2/CtIP protein creates a network of interactions with MRN that promotes the release of its auto-inhibition.
All these findings illustrate how an apparently disordered protein can lift the auto-inhibition of a nuclease and thus control the switch between different repair pathways, which is important in humans for cell cycle progression and meiosis.
More information: https://www.sciencedirect.com/science/article/pii/S1097276524004428?via%3Dihub
Contact: Raphaël GUEROIS raphael.guerois@i2bc.paris-saclay.fr
Sulfur starvation-induced autophagy in Saccharomyces cerevisiae involvesSAM-dependent signaling and transcription activator Met4
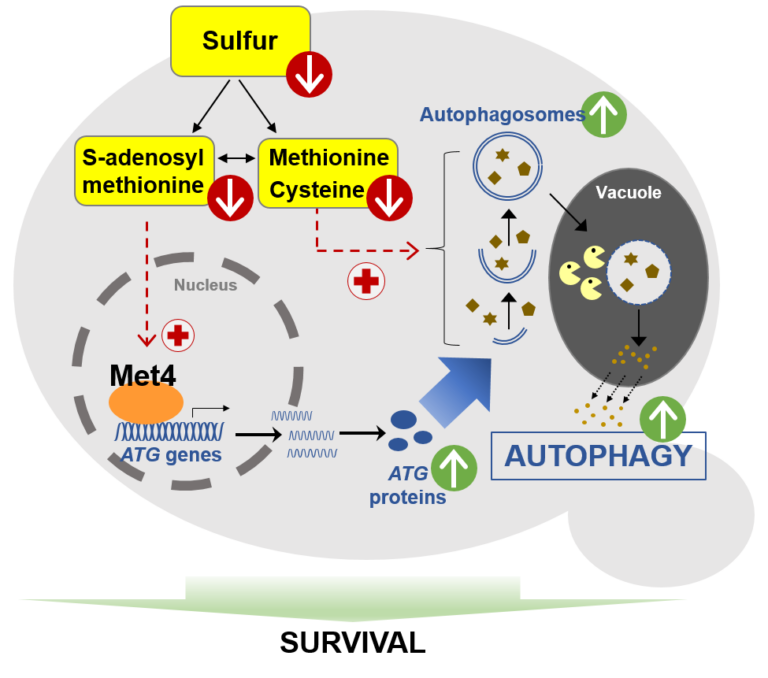
Unveiling a new property of bacteriophage-host interaction in the Autophagy has a key protective role under nutrient stresses. The authors unveil in yeast an original mechanism of induction of autophagy under sulfur deprivation that requires the transcription activator Met4 and involves S-adenosylmethionine as signaling molecule.
Autophagy is a key lysosomal degradative mechanism allowing a prosurvival response to stresses, especially nutrient starvation. Here we investigate the mechanism of autophagy induction in response to sulfur starvation in Saccharomyces cerevisiae. We found that sulfur deprivation leads to rapid and widespread transcriptional induction of autophagy-related (ATG) genes in ways not seen under nitrogen starvation. This distinctive response depends mainly on the transcription activator of sulfur metabolism Met4. Consistently, Met4 is essential for autophagy under sulfur starvation. Depletion of either cysteine, methionine or SAM induces autophagy flux. However, only SAM depletion can trigger strong transcriptional induction of ATG genes and a fully functional autophagic response. Furthermore, combined inactivation of Met4 and Atg1 causes a dramatic decrease in cell survival under sulfur starvation, highlighting the interplay between sulfur metabolism and autophagy to maintain cell viability. Thus, we describe a pathway of sulfur starvation induced autophagy depending on Met4 and involving SAM as signaling sulfur metabolite.
More information: https://doi.org/10.1038/s41467-024-51309-6
Contact: Laurent KURAS laurent.kuras@i2bc.paris-saclay.fr
Prophage induction can facilitate the in vitro dispersal of multicellular Streptomyces structures

Unveiling a new property of bacteriophage-host interaction in the context of multicellular aggregate dynamics – A multidisciplinary study involving three departments from I2BC (Genomes, Microbiology, and Virology) and a team from MICALIS.
Streptomyces are renowned for their prolific production of specialized metabolites with applications in medicine and agriculture. These multicellular bacteria present a sophisticated developmental cycle, and play a key role in soil ecology. Little is known about the impact of Streptomyces-phage on bacterial physiology. In this study, we investigated the conditions governing the expression and production of ‘Samy’, a prophage found in Streptomyces ambofaciens ATCC 23877. This siphoprophage is produced simultaneously with the activation of other mobile genetic elements. Remarkably, the presence and production of Samy increases bacterial dispersal under in vitro stress conditions. Altogether, this study unveiled a new property of a bacteriophage infection in the context of multicellular aggregate dynamics.
More information: https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3002725
Contact: Stéphanie BURY-MONE stephanie.bury-mone@i2bc.paris-saclay.fr
TLN468 changes the pattern of tRNA used to read through premature termination codons in CFTR

TLN468 promotes PTC readthrough by preferentially incorporating specific tRNAs based on the mutation.
Nonsense mutations account for 12 % of cystic fibrosis (CF) cases. The presence of a premature termination codon (PTC) leads to gene inactivation, which can be countered by the use of drugs stimulating PTC readthrough, restoring production of the full-length protein. We recently identified a new readthrough inducer, TLN468, more efficient than gentamicin.
We measured the readthrough induced by these two drugs with different cystic fibrosis transmembrane conductance regulator (CFTR) PTCs. We then determined the amino acids inserted at the S1196X, G542X, W846X and E1417X PTCs of CFTR during readthrough induced by gentamicin or TLN468. TLN468 significantly promoted the incorporation of one specific amino acid, whereas gentamicin did not greatly modify the pro- portions of the various amino acids incorporated relative to basal conditions. The function of the engineered missense CFTR channels corresponding to these four PTCs was assessed with and without potentiator. For the recoded CFTR, except for E1417Q and G542W, the PTC readthrough induced by TLN468 allowed the expression of CFTR variants that were correctly processed and had significant activity that was enhanced by CFTR modu- lators. These results suggest that it would be relevant to assess the therapeutic benefit of TLN468 PTC sup- pression in combination with CFTR modulators in preclinical assays.
More information: https://www.cysticfibrosisjournal.com/article/S1569-1993(24)00802-6/fulltext
Contact: Olivier NAMY olivier.namy@i2bc.paris-saclay.fr
A tumor suppressor that generates subnucleosomes

Researchers from the I2BC, the CNRGH (CEA, National Center of Human Genomics Research), and the University of Edinburgh have revealed how the BAF chromatin remodeler changes the structure and accessibility of genetic material by producing subnucleosomal particles. These findings are published in Nature Structural and Molecular Biology.
Mutations in genes encoding subunits of the BAF complex (BRG1/BRM-associated factors, also known as the SWI/SNF complex) are associated with 20% of human cancers, making this complex the second most frequently mutated factor after the p53 tumor suppressor.
The human genome exists in cells in the form of chromatin, composed of nucleosomes that condense and protect the genetic material. The nucleosomes represent a physical barrier for transcription factors and molecular machinery that regulate gene expression. The BAF complex is known for controlling chromatin opening at gene transcription regulatory elements. Until now, it was believed that the role of the BAF complex was to dissociate the nucleosomes to allow the transcription factors to access DNA.
Using a method that allows them to differentiate histone-free DNA fragments from nucleosomes and histone-containing particles smaller than nucleosomes (subnucleosomes), researchers coordinated by Matthieu Gérard (I2BC) revealed that the BAF complex facilitates the recruitment of the master transcription factor OCT4 using two distinct strategies: i) by generating the well-known histone-free DNA regions, which OCT4 binds if its target DNA motif is present; and ii) by producing a new class of subnucleosomal particles containing 50 to 80 base pairs associated with histones.
They show that the subnucleosomes act as a recruitment platform for OCT4 independently of the presence of its DNA motif. They identify in this publication a molecular mechanism in which the interaction between OCT4 and the subnucleosomes leads to a spectacular expansion of the OCT4 genomic binding interval. This mechanism aims to project OCT4 activity in chromatin opening within a genomic interval up to one order of magnitude larger than the interval bound by OCT4 on histone-free DNA.
This study suggests two main determinants for recruiting transcription factors onto the mammalian genome: the genetic determinant, the transcription factor motif within histone-free DNA, and an epigenetic determinant, which corresponds to the subnucleosomes produced by the BAF complex.
More information: https://www.nature.com/articles/s41594-024-01344-0
Contact: Matthieu GERARD matthieu.gerard@i2bc.paris-saclay.fr
BRCA2 stabilizes DMC1 nucleoprotein filaments in meiosis

To repair programmed double-strand breaks in meiosis, the DNA repair protein BRCA2 binds to the recombinase DMC1 either monomeric or assembled on single-stranded DNA through two different interfaces, and stabilizes DMC1 nucleoprotein filaments.
The BReast CAncer type 2 susceptibility protein (BRCA2), a tumor suppressor mutated in breast, ovarian and prostate cancers, plays a major role in the repair of DNA double-strand breaks by homologous recombination, both in somatic cells and during meiosis. BRCA2 interacts with the ubiquitous recombinase RAD51 , as well as the meiotic recombinase DMC1, and facilitates their loading at double-strand break sites. BRCA2 interacts with these recombinases via FxxA and FxPP motifs (called A and P motifs, respectively). In a study published recently in the journal Nucleic Acids Research, scientists from the INTGEN team at the I2BC (Université Paris Saclay, CEA, CNRS, Gif-sur-Yvette) and the PROXIMA-1 beamline at the SOLEIL synchrotron (Université Paris Saclay) solved the crystal structure of the complex between a BRCA2 fragment containing a P motif (PhePP) and the DMC1 protein. Together with the team of A. Zelensky and R. Kanaar (Erasmus Medical Center, Rotterdam), they showed that A and P motifs bind to distinct sites on the ATPase domain of recombinases. The P motif interacts with a site that is accessible in the octamers of DMC1 and the nucleoprotein filaments formed by DMC1 loaded onto single-stranded DNA. Furthermore, in collaboration with scientists from the Institut Gustave Roussy (Université Paris Saclay), they revealed that this interaction involves two adjacent DMC1 protomers, thereby increasing the stability of the nucleoprotein filaments. These results help to explain why the region encoded by exons 12 to 14 of the BRCA2 gene (the PhePP motif being encoded by exon 14) is essential during meiotic homologous recombination in mice (work by the teams of A. Zelensky and W. Baarends, Erasmus Medical Center, Rotterdam, Netherlands).
More information: https://doi.org/10.1093/nar/gkae452
Contact: Simona MIRON <simona.miron@i2bc.paris-saclay.fr> & Sophie ZINN <sophie.zinn@i2bc.paris-saclay.fr>
SYNTERUPTOR: mining genomic islands for non-classical specialized metabolite gene clusters

In search of the hidden gene cluster: Synteruptor, a new tool for identifying bacterial genomic islands and exploring their content in the quest for new specialized metabolism gene clusters.
Microbial specialized metabolite biosynthetic gene clusters (SMBGCs) are a formidable source of natural products of pharmaceutical interest. With the multiplication of genomic data available, very efficient bioinformatic tools for automatic SMBGC detection have been developed. Nevertheless, most of these tools identify SMBGCs based on sequence similarity with enzymes typically involved in specialised metabolism and thus may miss SMBGCs coding for undercharacterised enzymes. In this article, we present Synteruptor (https://bioi2.www.i2bc.paris-saclay.fr/synteruptor), a program that identifies genomic islands, known to be enriched in SMBGCs, in the genomes of closely related species. With this tool, we identified a SMBGC in the genome of Streptomyces ambofaciens ATCC23877, undetected by antiSMASH, the well-known and most used tool for SMBGC identification, prior to antiSMASH 5. We experimentally demonstrated that this SMBGC directs the biosynthesis of two metabolites, one of which was identified as sphydrofuran. Synteruptor is also a valuable resource for the delineation of individual SMBGCs within antiSMASH regions that may encompass multiple clusters, and for refining the boundaries of these SMBGCs.
More information: https://doi.org/10.1093/nargab/lqae069
Contact: Sylvie LAUTRU <sylvie.lautru@i2bc.paris-saclay.fr> & Olivier LESPINET <olivier.lespinet@i2bc.paris-saclay.fr>
Acinetobacter baumannii satellite phage Aci01-2-Phanie depends on a helper myophage for its multiplication

Phanie is a phi-29 like phage that protects its genome inside non-infectious podovirus-like particles, requiring acquisition of the tail from a myovirus helper for production of infectious chimeras.
Viruses of bacteria (bacteriophages or phages) display a remarkable genomic diversity reflecting the complex interaction they have built with their host during evolution. In addition to fully infectious viral particles, different types of genetic elements were described to require a helper phage for production of infectious virions. The genome of these satellite parasite sequences may share sequences with phages or be more closely related to non-viral chromosome islands. We describe a new mode of satellite phage dependence on a helper phage. Phanie, like the genetically related Bacillus subtilis phage phi29, replicates its linear dsDNA by a protein primed-mechanism and protects it inside podovirus-like particles. However, these particles are defective, requiring acquisition of the tail from a myovirus helper for production of infectious virions. Formation of chimeras between a phi29-like podovirus and a helper contractile tail reveals an unexpected association between very different bacterial viruses.
More information: https://journals-asm-org.insb.bib.cnrs.fr/doi/10.1128/jvi.00667-24
Contact: Christine Pourcel christine.pourcel@i2bc.paris-saclay.fr
Gradual ER calcium depletion induces a progressive and reversible UPR signaling

The UPR sensors show high plasticity in sensing physiological alterations by closely reporting the variations in ER Ca2+ levels. Through this mechanisms cells could rapidly adapt stress response signaling pathways to variations in ER homeostasis.
The unfolded protein response (UPR) is a widespread signal transduction pathway triggered by endoplasmic reticulum (ER) stress. Because calcium (Ca2+) is a key factor in the maintenance of ER homeostasis, massive Ca2+ depletion of the ER is a potent inducer of ER stress. Although moderate changes in ER Ca2+ drive the ubiquitous Ca2+ signaling pathways, a possible incremental relationship between UPR activation and Ca2+ changes has yet to be described. Here, we determine the sensitivity and time-dependency of activation of the three ER stress sensors, IRE1α, PERK and ATF6α in response to controlled changes in the concentration of ER Ca2+ in human cultured cells. Combining Ca2+ imaging, FRAP experiments, biochemical analyses and mathematical modeling, we uncover a non-linear rate of activation of the IRE1α branch of UPR, as compared to the PERK and ATF6α branches that become activated gradually with time and are sensitive to more important ER Ca2+ depletions. However, the three arms are all activated within a 1h timescale. The model predicted the deactivation of PERK and IRE1α upon refilling the ER with Ca2+. Accordingly, we showed that ER Ca2+ replenishment leads to the complete reversion of IRE1α and PERK phosphorylation in less than 15 min, thus revealing the highly plastic character of the activation of the upstream UPR sensors. In conclusion, our results reveal a dynamic and dose-sensitive Ca2+-dependent activation/deactivation cycle of UPR induction, which could tightly control cell fate upon acute and/or chronic stress.
More information: https://doi.org/10.1093/pnasnexus/pgae229
Contact: Ilaria Pontisso (ilaria.pontisso@i2bc.paris-saclay.fr)
Timing is success! Bacteriophage tail completion proteins are essential regulators of viral DNA delivery to host bacteria

Researchers at I2BC and their collaborators have conducted an extensive functional analysis of a highly conserved Tail Completion Protein (TCP) in the assembly and infectivity of tailed bacteriophages.
Their investigation highlights the role of bacteriophage SPP1 TCP gp16.1 as a structural element of the tail, serving two distinct purposes. Firstly, gp16.1 assists assembly of the tail interface that binds to the phage capsid. Secondly, it ensures proper delivery of phage DNA to the bacterial cytoplasm.
In the absence of gp16.1, assembled viral particles are undistinguishable from wild-type virions and eject DNA efficiently in vitro. However, upon interaction with the host bacteria, they release their DNA into the extracellular space. Transfer of DNA from the viral particle to the bacterial cytoplasm requires prior localized digestion of the bacterial cell wall, likely facilitated by the tail tip, and the formation of a lipophilic channel in the bacterial membrane for DNA passage. Successful infection depends on precise timing, involving phage binding to the receptor, creating a pathway for DNA passage through the cell envelope, and DNA exit from the particle into the bacterial cytoplasm.
The authors propose that the gp16.1 critical function is achieved by its positioning at the tail end proximal to the capsid to form a complex with the Tape Measure Protein (TMP) and the Tail Tube Protein (TTP). These interactions conceivably regulate the timing of TMP and DNA release. This could result of narrowing the internal diameter of the tail tube to retain the TMP, slowing down release of the TMP and DNA until a continuous hydrophilic channel forms between the tail and the bacterial cytoplasm. Their study unravels the dual role of gp16.1 in tail assembly and in delivery of viral DNA into bacteria. Both functions are most likely exerted by the large superfamily of TCPs that are essential components of phages with long tails.
More information: https://www.nature.com/articles/s42003-024-06221-6
Contact: Isabelle Auzat isabelle.auzat@i2bc.paris-saclay.fr
Mechanism of DNA unwinding by MCM8-9 in complex with HROB

Researchers from the AMIG team (I2BC department), in collaboration with the IRB (Switzerland), have modeled the interaction between HROB and the helicases MCM8-MCM9, some mutations of which predispose individuals to infertility or cancer. They demonstrate that HROB promotes the catalytic activity of the MCM8-MCM9 complex but does not play a role in its recruitment or stability.
The proteins MCM8 and MCM9 have recently been discovered as involved in multiple processes, normal and pathological, related to DNA replication, meiosis, homologous recombination, and mismatch repair. These proteins are helicases that have the ability to move along DNA and separate the two DNA strands. Variants of these proteins may predispose carriers to disorders such as infertility and cancer. In 2019, a third partner, HROB, was identified as associated with these two helicases without its mechanisms of action being understood. Since HROB is capable of interacting with DNA, three functional hypotheses can be considered:
1) HROB participates in the recruitment of helicases on DNA,
2) HROB stabilizes the assembly of MCM8 and MCM9 on DNA,
3) HROB promotes the catalytic activity of pre-assembled helicases.
In a study published in the journal Nature Communications, researchers from the AMIG team at the Institute of Integrative Biology of the Cell (I2BC) collaborated with a team from the Institute of Research in Biomedicine (IRB) in Switzerland to establish different structural models of the MCM8-MCM9-HROB complex. Guided by these models, a set of simple and compensatory mutations allowed the dissection of the functional role of HROB. The teams showed that only the third hypothesis was correct and that HROB is an essential factor for activating the MCM8-MCM9 helicase but not for its recruitment or stability. The structural model suggests that by transiently altering the conformation of a MCM9 subunit, HROB stimulates the translocation of the helicase along the DNA. Based on this model, it was even possible to design mutations that increase the efficiency of the helicase in the presence of HROB.
More information: https://www.nature.com/articles/s41467-024-47936-8
Contact: Raphaël Guerois raphael.guerois@i2bc.paris-saclay.fr
Nuclear receptor signaling via NHR-49/MDT-15 regulates stress resilience and proteostasis in response to reproductive and metabolic cues

The production of unhealthy eggs leads to improved maternal stress resilience and proteostasis in C. elegans. This study identifies the nuclear hormone receptor NHR-49 as a critical regulator that is activated by elevated fat stores and acts by potentiating the heat shock response.
The ability to sense and respond to proteotoxic insults declines with age, leaving cells vulnerable to chronic and acute stressors. Reproductive cues modulate this decline in cellular proteostasis to influence organismal stress resilience in Caenorhabditis elegans We previously uncovered a pathway that links the integrity of developing embryos to somatic health in reproductive adults. Here, we show that the nuclear receptor NHR-49, an ortholog of mammalian peroxisome proliferator-activated receptor α (PPARα), regulates stress resilience and proteostasis downstream from embryo integrity and other pathways that influence lipid homeostasis and upstream of HSF-1. Disruption of the vitelline layer of the embryo envelope, which activates a proteostasis-enhancing intertissue pathway in somatic cells, triggers changes in lipid catabolism gene expression that are accompanied by an increase in fat stores. NHR-49, together with its coactivator, MDT-15, contributes to this remodeling of lipid metabolism and is also important for the elevated stress resilience mediated by inhibition of the embryonic vitelline layer. Our findings indicate that NHR-49 also contributes to stress resilience in other pathways known to change lipid homeostasis, including reduced insulin-like signaling and fasting, and that increased NHR-49 activity is sufficient to improve proteostasis and stress resilience in an HSF-1-dependent manner. Together, our results establish NHR-49 as a key regulator that links lipid homeostasis and cellular resilience to proteotoxic stress.
More information: https://genesdev.cshlp.org/content/early/2024/05/30/gad.351829.124
Contact: Ambre Sala ambre.sala@i2bc.paris-saaclay.fr
Staphylococcus aureus in the battle for iron:
three post-transcriptional regulations at work

A feed-forward loop controlled by the regulatory sRNA IsrR and the aconitase moonlighting activity prevent aconitase production during iron deficiency.
Staphylococcus aureus, responsible for nosocomial infections and septicemia, is the leading cause of bacterial mortality in most countries of the world. Its pathogenicity relies on its adaptation to a wide range of biotopes.
As iron bioavailability is sometimes a limiting factor for growth, bacteria have developed strategies to scavenge iron and reduce its utilization under iron-depleted growth conditions. One such strategy involves the modulation of iron-utilizing proteins through the action of non-coding regulatory RNAs (sRNA). Notably, our previous research unveiled the significance of IsrR, a staphylococcal sRNA required for optimal growth in iron-depleted media and sustaining full virulence (Coronel-Tellez, 2022). Triggered by iron starvation, IsrR coordinates the down-regulation of genes encoding [Fe-S]-containing enzymes, including aconitase.
Aconitase converts citrate to isocitrate. CcpE, a citrate-activated transcriptional regulator, positively regulates its gene. In this new work, we show that IsrR inhibits the translation of aconitase mRNA and of ccpE mRNA, thus establishing a coherent feed-forward regulatory loop. This dual mechanism of repression ensures effective control of aconitase production by IsrR. Aconitase is a protein known for its second (moonlighting) activity in the absence of its [Fe-S] cluster; it becomes an RNA-binding protein (RBP) with regulatory capacity. This characteristic is conserved from prokaryotes to eukaryotes. Here, we show that the RBP activity of S. aureus aconitase reduces the amount of aconitase and influences the expression of genes encoding metabolic enzymes during iron deficiency.
Our study reveals the complex network of post-transcriptional regulations governing aconitase production that enables S. aureus to thrive in iron-deficient conditions. Importantly, these conditions are encountered by bacteria in the host as a result of a defense response known as nutritional immunity, in which iron and other essential nutrients are sequestered to prevent infection.
More information: https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkae506/7692338
Contact: Philippe Bouloc philippe.bouloc@i2bc.paris-saclay.fr
Hundreds of antimicrobial peptides create a selective barrier for insect gut symbionts

Antimicrobial peptides shape the gut microbiota biogeography in insects.
The microbioata is usually not homogeneously dispersed in the animal gut but spatially structured in microenvironments. The microbiota in the gut of the bean bug Riptortus pedestris displays a sharp divide between the anterior and posterior midgut with a multispecies bacterial community in the anterior region and a specific, mono-species Caballeronia symbiont population in the posterior region. In this collaborative work between I2BC teams, the Next-generation sequencing plateform, the scanning electron microscopy plateform of MICALIS and a team from the AIST, Sapporo Japan, we found that this insect deploys in the midgut an arsenal of several hundreds of antimicrobial peptides. These peptides have antimicrobial activity against diverse bacteria but posterior midgut symbionts have elevated resistance while mutants of these symbionts in resistance genes have reduced capacity to colonize the posterior midgut. The peptides create thus a selective environment restricting the type of bacteria from the anterior midgut microbiota that have a chance to establish in the posterior midgut. This finding highlights a mechanism that contributes in the construction of an exclusive niche for beneficial gut symbionts.
More information: https://www.pnas.org/doi/10.1073/pnas.2401802121
Contact: Peter Mergaert peter.mergaert@i2bc.paris-saclay.fr
Imprinting gene regulation:
Importance of a long non-coding RNA
and of DNA methylation levels at an essential CTCF binding site
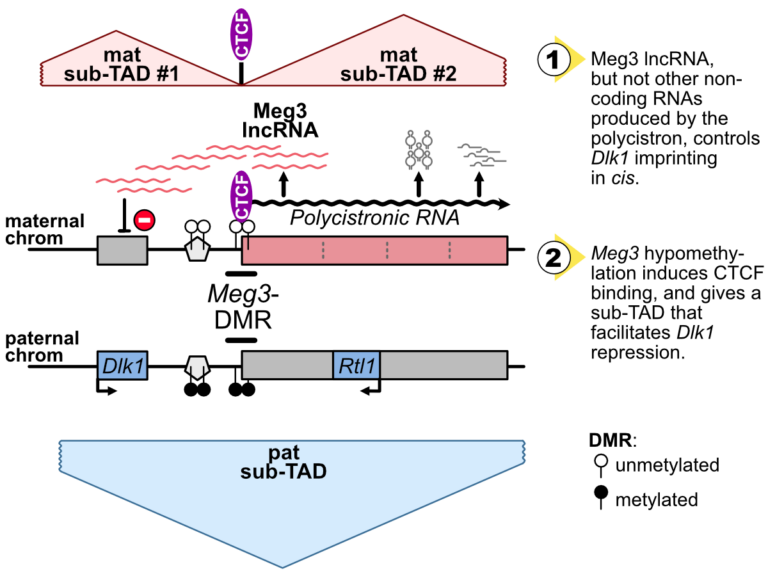
The Dlk1-Meg3-Dio3 domain is imprinted. Scientists at the IGMM and the I2BC have determined that Meg3 long non-coding RNA, as well as DNA methylation levels at an essential CTCF binding site in the domain, control Dlk1 imprinting in cis.
The imprinted Dlk1-Dio3 domain comprises the Dlk1 and Rtl1 developmental genes that are solely expressed from the paternal chromosome, and a maternally-expressed polycistron that is further processed into the Meg3 long non-coding RNA (lncRNA) and many other small non-coding RNAs.
In collaborative work between scientists at the IGMM (R. Feil’s group; Montpellier) and at the I2BC (D. Noordermeer’s group; Gif-sur-Yvette), researchers studied the precise role of Meg3 lncRNA and of its promoter for the control of Dlk1 and Rtl1 imprinted gene expression. Using mutant cells with either premature termination of transcription or deletion of part of the polycistron, they found that Meg3 lncRNA, but not other non-coding RNAs produced by the polycistron, controls Dlk1 imprinting in cis.
The maternal expression of this polycistron is driven by the Meg3 differentially methylated region (Meg3-DMR) which is unmethylated on the maternal allele and acts there as an active promoter. Besides, the Meg3-DMR includes a known binding site for the architectural CTCF protein, that binds to the unmethylated maternal allele only. In this study, the researcher showed that the maternal Dlk1-Dio3 locus is organized into sub-Topologically Associating domains (sub-TADs) that are hinged by the allelic binding of CTCF to the maternal Meg3-DMR. They further found that the methylation levels at the Meg3-DMR are instructive for CTCF binding and thereby dictate distinctive sub-TAD organization at the two parental alleles which, in turns, facilitates Dlk1 repression on the maternal allele.
Altogether, this collaborative work revealed that, the maternally-expressed Meg3 lncRNA controls Dlk1 silencing in cis, and that the low methylation levels at the maternal Meg3-DMR allows for specific sub-TADs structuration that concurrently contributes to Dlk1 silencing.
More information: https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkae247/7645245?searchresult=1
Contact: Benoit Moindrot benoit.moindrot@i2bc.paris-saclay.fr
Uncoupling programmed DNA cleavage and repair
scrambles the Paramecium somatic genome
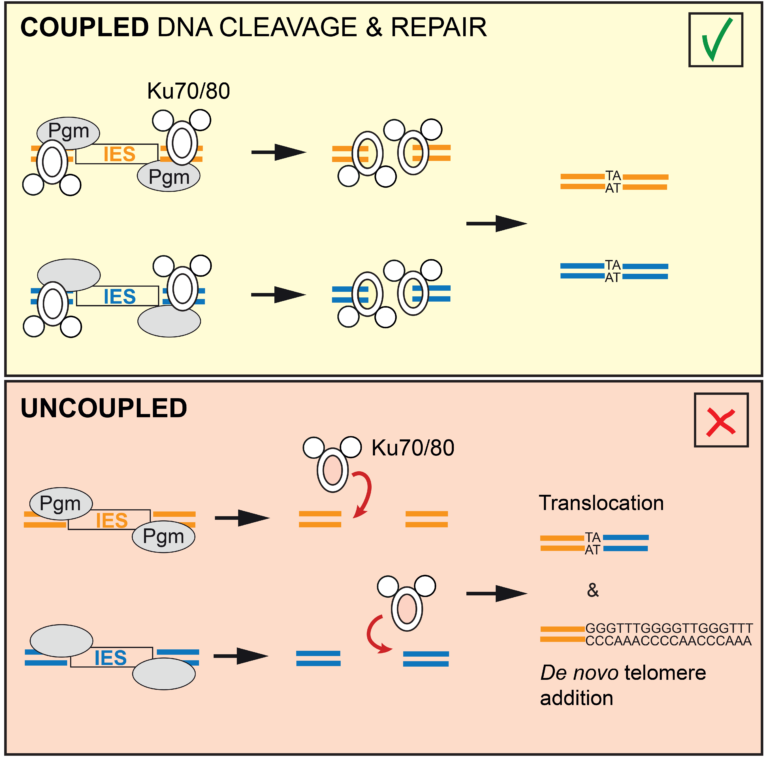
No Ku – no cut: how the ciliate Paramecium tetraurelia protects its somatic genome from translocations and aberrant chromosome fragmentation during developmentally programmed rearrangements.
DNA double-strand breaks (DSBs) are a major threat to genome integrity. When incorrectly repaired, they can lead to genome rearrangements, chromosome instability, diseases, or cell death. Yet, key physiological processes, such as antibody gene assembly or meiotic recombination, rely on DSB induction. How living organisms cope with programmed DSBs is a major question in genome biology. The “Programmed genome rearrangements” team studies Paramecium to dissect developmentally programmed DNA elimination mechanisms. During each sexual cycle, tens of thousands of germline Internal Eliminated Sequences (IESs), scattered all along chromosomes, undergo precise excision. This process requires DSB introduction at IES boundaries, followed by error-free stitching of flanking DNA fragments by the non-homologous end joining (NHEJ) pathway. The team previously reported that the presence of a specialized variant of the NHEJ factor Ku is a pre-requisite for the activation of DNA cleavage, indicating that the two steps of the reaction are coupled. In this study, they engineered a DNA binding-deficient Ku mutant, which they characterized in collaboration with the IntGen team from the B3S department. They showed that the mutant Ku activates DNA cleavage, but is defective in repair. Unrepaired broken ends at IES boundaries are then trimmed and healed by telomere addition. By co-expressing wild-type Ku with the mutant, they were able to uncouple the cleavage and repair steps during IES excision. Using high throughput DNA sequencing and novel dedicated bioinformatic tools, they showed that chromosome translocations were largely favored under these conditions. This work demonstrates that coupling DNA cleavage and DSB repair ensures faithful genome assembly during programmed rearrangements.
More information: https://www.cell.com/cell-reports/fulltext/S2211-1247(24)00329-2
Contact: Mireille Bétermier mireille.betermier@i2bc.paris-saclay.fr – Julien Bischerour – julien.bischerour@i2bc.paris-saclay.fr
Transcription-induced DNA supercoiling clears RNA polymerase’s path in bacterial chromatin

Positive DNA supercoiling generated ahead of RNA polymerase during transcription elongation dislodges nucleoid-structuring protein H-NS from the DNA.
Bacterial chromosomal DNA is condensed more than 1000 times inside the cell. This condensation results from a slight underwinding of the double helix, leading to the formation of loops of negatively supercoiled DNA (plectonemes) and from interactions with structural proteins that help compact these loops. However, these chromatin-associated proteins obstruct transcription and need to be transiently removed to allow gene expression. This is the case for H-NS, an abundant protein that blocks the expression of numerous genes. In Gram-negative pathogenic bacteria such as Salmonella, H-NS represses the expression of the majority of virulence genes. Displacement of H-NS becomes necessary during the infection process, which relies on the function of virulence gene products.
In a study published in the journal Nature Communications, researchers from the Genome Biology department of the Institute for Integrative Biology of the Cell (I2BC) present evidence that binding of H-NS to DNA is destabilized, from a distance, by the positive supercoils forming ahead of approaching RNA polymerase. It is known that the process of transcription induces DNA supercoiling by forcing the rotation of the DNA axis during elongation. To explain the destabilization of H-NS:DNA complexes by positive supercoiling, the researchers propose a model in which H-NS oligomers play a scaffolding role in negatively supercoiled DNA by bridging opposite arms of the plectonemic structure. Accumulation of positive supercoils and concomitant DNA axis rotation ahead of the approaching RNA polymerase cause the H-NS-bound, negatively supercoiled plectoneme to unroll, leading to the collapse of the scaffold and allowing RNA polymerase to continue on its path.
More information: https://www.nature.com/articles/s41467-024-47114-w
Contact: Lionelo Bossi lionello.bossi@i2bc.paris-saclay.fr
A Fatty Acid Anabolic Pathway in Specialized-Cells Sustains a Remote Signal that Controls Egg Activation in Drosophila
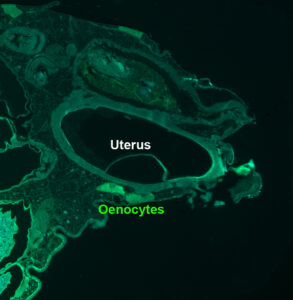
While egg activation, i.e. oocyte to embryo transition, was not known to depend on physiological non-genital signal, this study shows that in Drosophila it depends on a very-long-chain-fatty-acid produced in the oenocytes (lipid metabolism specialized cells).
Egg activation, representing the critical oocyte-to-embryo transition, provokes meiosis completion, modification of the vitelline membrane to prevent polyspermy, and translation of maternally provided mRNAs. This transition is triggered by a calcium signal induced by spermatozoon fertilization in most animal species, but not in insects. In Drosophila melanogaster, mature oocytes remain arrested at metaphase-I of meiosis and the calcium-dependent activation occurs while the oocyte moves through the genital tract. Here, we discovered that the oenocytes of fruitfly females are required for egg activation. Oenocytes, cells specialized in lipid-metabolism, are located beneath the abdominal cuticle. In adult flies, they synthesize the fatty acids (FAs) that are the precursors of cuticular hydrocarbons (CHCs), including pheromones. The oenocyte-targeted knockdown of a set of FA-anabolic enzymes, involved in very-long-chain fatty acid (VLCFA) synthesis, leads to a defect in egg activation. Given that some but not all of the identified enzymes are required for CHC/pheromone biogenesis, this putative VLCFA-dependent remote control may rely on an as-yet unidentified CHC or may function in parallel to CHC biogenesis. Additionally, we discovered that the most posterior ventral oenocyte cluster is in close proximity to the uterus. Since oocytes dissected from females deficient in this FA-anabolic pathway can be activated in vitro, this regulatory loop likely operates upstream of the calcium trigger. To our knowledge, our findings provide the first evidence that a physiological extra-genital signal remotely controls egg activation. Moreover, our study highlights a potential metabolic link between pheromone-mediated partner recognition and egg activation.
More information: https://doi.org/10.1371/journal.pgen.1011186
Contact: Jacques Montagne jacques.montagne@i2bc.paris-saclay.fr
A highly conserved ligand-binding site for AccA transporters of antibiotic and quorum-sensing regulator in Agrobacterium leads to a different specificity
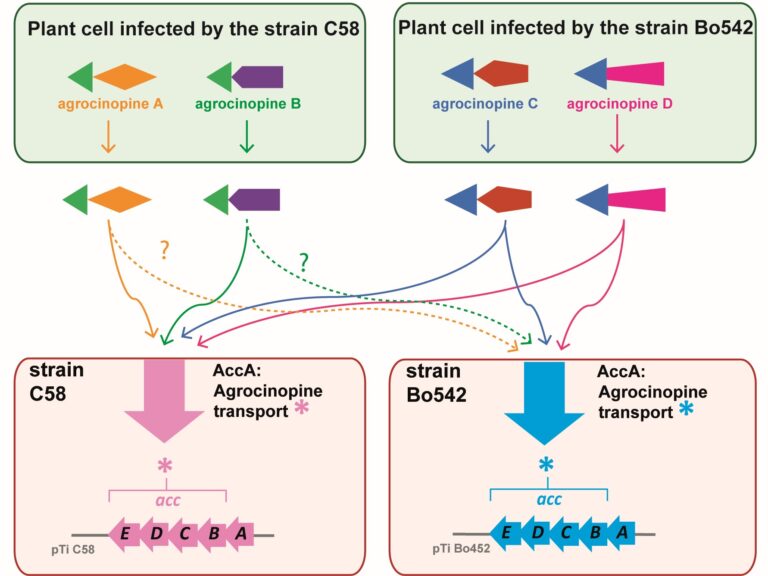
The highly conserved ligand binding site of the AccA transporters of antibiotic and quorum-sensing regulator in Agrobacterium is not linked to a conserved specificity.
Plants genetically modified by the pathogenic Agrobacterium strain C58 synthesize agrocinopines A and B, whereas those modified by the pathogenic strain Bo542 produce agrocinopines C and D. The four agrocinopines (A, B, C and D) serve as nutrients by agrobacteria and signaling molecule for the dissemination of virulence genes. They share the uncommon pyranose-2-phosphate motif, represented by the L-arabinopyranose moiety in agrocinopines A/B and the D-glucopyranose moiety in agrocinopines C/D, also found in the antibiotic agrocin 84. They are imported into agrobacterial cytoplasm via the Acc transport system, including the solute-binding protein AccA coupled to an ABC transporter. We have previously shown that unexpectedly, AccA from strain C58 (AccAC58) recognizes the pyranose-2-phosphate motif present in all four agrocinopines and agrocin 84, meaning that strain C58 is able to import agrocinopines C/D, originating from the competitor strain Bo542. Here, using agrocinopine derivatives and combining crystallography, affinity and stability measurements, modeling, molecular dynamics, in vitro and vivo assays, we show that AccABo542 and AccAC58 behave differently despite 75% sequence identity and a nearly identical ligand binding site. Indeed, strain Bo542 imports only compounds containing the D-glucopyranose-2-phosphate moiety, and with a lower affinity compared to strain C58. This difference in import efficiency makes C58 more competitive than Bo542 in culture media. We can now explain why Agrobacterium/Allorhizobium vitis strain S4 is insensitive to agrocin 84, although its genome contains a conserved Acc transport system. Overall, our work highlights AccA proteins as a case study, for which stability and dynamics drive specificity.
More information: https://portlandpress.com/biochemj/article-abstract/481/2/93/233802/A-highly-conserved-ligand-binding-site-for-AccA?redirectedFrom=fulltext
Contact: Solange Morera solange.morera@i2bc.paris-saclay.fr
Epigenetics: combining flexible and rigid regions into a single structure to ensure genome replication and stability

The AMIG team at I2BC, in collaboration with teams from the Institut Curie and the Synchrotron Soleil, shows that the CAF-1 protein combines in its spatial organization flexible regions and rigid modules to deposit histones on DNA and effectively couple this process to DNA synthesis.
Genome and epigenome integrity in eukaryotes depends on the proper coupling of histone deposition with DNA synthesis. This process relies on the evolutionary conserved histone chaperone CAF-1 for which the links between structure and functions are still a puzzle. While studies of the Saccharomyces cerevisiae CAF-1 complex enabled to propose a model for the histone deposition mechanism, we still lack a framework to demonstrate its generality and in particular, how its interaction with the polymerase accessory factor PCNA is operating. Here, we reconstituted a complete SpCAF-1 from fission yeast. We characterized its dynamic structure using NMR, SAXS and molecular modeling together with in vitro and in vivo functional studies on rationally designed interaction mutants. Importantly, we identify the unfolded nature of the acidic domain which folds up when binding to histones. We also show how the long KER helix mediates DNA binding and stimulates SpCAF-1 association with PCNA. Our study highlights how the organization of CAF-1 comprising both disordered regions and folded modules enables the dynamics of multiple interactions to promote synthesis-coupled histone deposition essential for its DNA replication, heterochromatin maintenance, and genome stability functions.
More information: https://elifesciences.org/articles/91461
Contact: Françoise Ochsenbein francoise.ochsenbein@i2bc.paris-saclay.fr
A genome-wide comprehensive analysis of nucleosome positioning in yeast

We show that the regulation and compartmentalisation of nucleosomal organisation require the concomitant actions of local and global processes that are maintained actively by energy consuming factors.
In eukaryotic cells, the one-dimensional DNA molecules need to be tightly packaged into the spatially constraining nucleus. Folding is achieved on its lowest level by wrapping the DNA around nucleosomes. Their arrangement regulates other nuclear processes, such as tran- scription and DNA repair. Despite strong efforts to study nucleosome positioning using Next Generation Sequencing (NGS) data, the mechanism of their collective arrangement along the gene body remains poorly understood. Here, we classify nucleosome distributions of protein-coding genes in Saccharomyces cerevisiae according to their profile similarity and analyse their differences using functional Principal Component Analysis. By decomposing the NGS signals into their main descriptive functions, we compared wild type and chromatin remodeler-deficient strains, keeping position-specific details preserved whilst considering the nucleosome arrangement as a whole. A correlation analysis with other genomic proper- ties, such as gene size and length of the upstream Nucleosome Depleted Region (NDR), identified key factors that influence the nucleosome distribution. We reveal that the RSC chromatin remodeler—which is responsible for NDR maintenance—is indispensable for decoupling nucleosome arrangement within the gene from positioning outside, which inter- fere in rsc8-depleted conditions. Moreover, nucleosome profiles in chd1Δ strains displayed a clear correlation with RNA polymerase II presence, whereas wild type cells did not indicate a noticeable interdependence. We propose that RSC is pivotal for global nucleosome orga- nisation, whilst Chd1 plays a key role for maintaining local arrangement.
More information: https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1011799
Contact: Arach GOLDAR <arach.goldar@i2bc.paris-saclay.fr>
Activity of MukBEF for chromosome management in E. coli
and its inhibition by MatP

The MukBEF condensin of E. coli loads at the replication fork and is unloaded at the Ter region by MatP.
Structural maintenance of chromosomes (SMC) complexes share conserved structures and serve a common role in maintaining chromosome architecture. In the bacterium Escherichia coli, the SMC complex MukBEF is necessary for rapid growth and the accurate segregation and positioning of the chromosome, although the specific molecular mechanisms involved are still unknown. Here, we used a number of in vivo assays to reveal how MukBEF controls chromosome conformation and how the MatP/matS system prevents MukBEF activity. Our results indicate that the loading of MukBEF occurs preferentially on newly replicated DNA, at multiple loci on the chromosome where it can promote long-range contacts in cis even though MukBEF can promote long-range contacts in the absence of replication. Using Hi-C and ChIP-seq analyses in strains with rearranged chromosomes, the prevention of MukBEF activity increases with the number of matS sites and this effect likely results from the unloading of MukBEF by MatP. Altogether, our results reveal how MukBEF operates to control chromosome folding and segregation in E. coli.
More information: https://elifesciences.org/articles/91185
Contact: Stéphane DUIGOU <stephane.duigou@i2bc.paris-saclay.fr>
Cooperation between two modes for DNA replication initiation in the archaeon Thermococcus barophilus
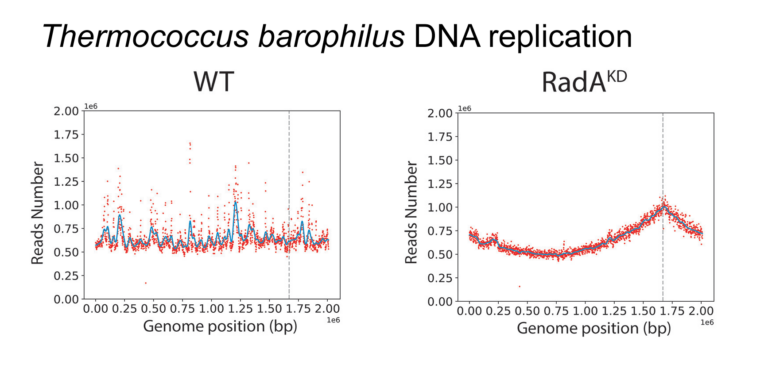
Demonstration that diverse physiological states influence the mode of DNA replication initiation in the archaeon Thermococcus.
The mechanisms underpinning the replication of genomic DNA have recently been challenged in Archaea. Indeed, the lack of origin of replication has no deleterious effect on growth, suggesting that replication initiation relies on homologous recombination. Recombination-dependent replication (RDR) appears to be based on the recombinase RadA, which is of absolute requirement when no initiation origins are detected. The origin of this flexibility in the initiation of replication and the extent to which it is used in nature are yet to be understood. We combined deep sequencing and genetics to elucidate the dynamics of oriC utilization according to growth phases. We discovered that in Thermococcus barophilus, the use of oriC diminishes from the lag to the middle of the log phase, and subsequently increases gradually upon entering the stationary phase. Although oriC demonstrates no indispensability, RadA does exhibit essentiality. Notably, a knockdown mutant strain provides confirmation of the pivotal role of RadA in RDR for the first time. Thus, we demonstrate the existence of a tight combination between oriC utilization and homologous recombination to initiate DNA replication along the growth phases. Overall, this study demonstrates how diverse physiological states can influence the initiation of DNA replication, offering insights into how environmental sensing might impact this fundamental mechanism of life.
More information: https://pubmed.ncbi.nlm.nih.gov/38421162/
Contact: Jacques OBERTO <jacques.oberto@i2bc.paris-saclay.fr>
JAK-STAT-dependent contact between follicle cells and the oocyte controls Drosophila anterior-posterior polarity and germline development
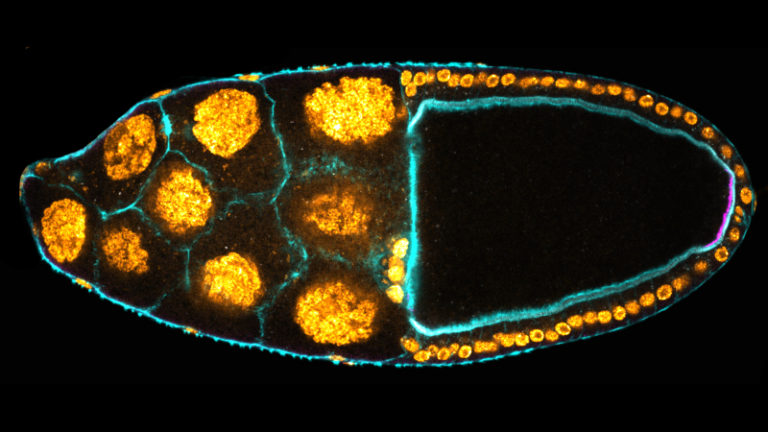
The authors identified a population of somatic cells in Drosophila follicles that elaborate filopodia penetrating the oocyte, thereby maintaining the contact between the two tissues. This is essential to control polarity and germline development of the future embryo.
The number of embryonic primordial germ cells is determined, in Drosophila, by the quantity of germ plasm, whose assembly starts in the posterior region of the oocyte during oogenesis. Here, we report that extending JAK-STAT activity in the posterior somatic follicular epithelium leads to an excess of primordial germ cells in the future embryo. We show that JAK-STAT signaling is necessary for the differentiation of approximately 20 specialized follicle cells maintaining tight contact with the oocyte. These cells define, in the underlying posterior oocyte cortex, the anchoring of the germ cell determinant oskar mRNA. We reveal that the apical surface of these posterior anchoring cells extends long filopodia penetrating the oocyte. We identify two JAK-STAT targets in these cells that are each sufficient to extend the zone of contact with the oocyte, thereby leading to production of extra primordial germ cells. JAK-STAT signaling thus determines a fixed number of posterior anchoring cells required for anterior-posterior oocyte polarity and for the development of the future germline.
More information: https://doi-org.insb.bib.cnrs.fr/10.1038/s41467-024-45963-z
Contact: Marianne MALARTRE <marianne.malartre@i2bc.paris-saclay.fr>
Transposon sequencing reveals genes enabling insect gut colonization by the symbiont Caballeronia insecticola

High-througput genetic screens with Tn-seq in an insect gut bacterium reveals gut-derived nutrients consumed by the symbiont.
Caballeronia insecticola is a bacterium belonging to the Burkholderia sensu lato, able to colonize multiple environments like soils and the gut of the bean bug Riptortus pedestris. We constructed a saturated Himar1 mariner transposon library and revealed by transposon-sequencing (Tn-seq) that 498 protein-coding genes constitute the essential genome of C. insecticola for growth in free-living conditions. By comparing essential gene sets of C. insecticola and seven related Burkholderia s.l. strains, only 120 common genes were identified indicating that a large part of the essential genome is strain-specific. In order to reproduce specific nutritional conditions that are present in the gut of R. pedestris, we grew the mutant library in minimal media supplemented with candidate gut nutrients and identified several condition-dependent fitness-defect genes by Tn-seq. To validate the robustness of the approach, insertion mutants in six fitness genes were constructed and their growth-deficiency in media supplemented with the corresponding nutrient was confirmed. The mutants were further tested for their efficiency in R. pedestris gut colonization, confirming that gluconeogenic carbon sources, taurine and inositol, are nutrients consumed by the symbiont in the gut. Thus, our study provides insights about specific contributions provided by the insect host to the bacterial symbiont.
More information: https://doi.org/10.1093/ismeco/ycad001
Contact: Peter MERGAERT <peter.mergaert@i2bc.paris-saclay.fr>
Protein-protein interactions: how to push forward the limits of the revolutionary AlphaFold2 programme?
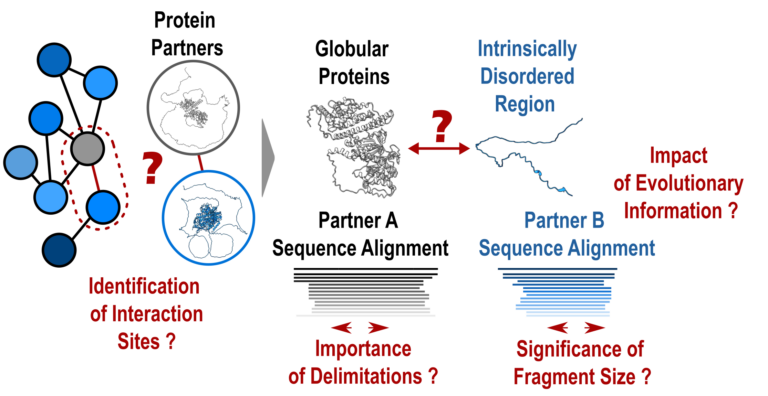
AlphaFold2 is revolutionising protein structure prediction and structural biology practices. However, it may prove less effective for certain protein assemblies, particularly when they depend on intrinsically disordered regions. In an article published in Nature Communications, researchers from the I2BC show that applying a fragmentation strategy to the protein partners of such assemblies very significantly improves AlphaFold2’s prediction capacity.
Mapping protein-protein interaction networks is essential for understanding the dynamics of cellular functions and their cross-regulation. Precise knowledge of interaction sites makes it possible to specifically perturb the proteins in these networks and understand the synergies and competitions that ensure cell function.
Unfortunately, a great amount of structural information is still lacking to provide a detailed understanding of the organisation of interaction networks. The AlphaFold2 artificial intelligence programme has demonstrated a remarkable ability to predict the structures of protein assemblies that have co-evolved over long time scales. Its performance remained poorly characterised for assemblies involving intrinsically disordered regions, which often mediate transient and dynamic interactions during evolution.
In a study published in the journal Nature Communications, researchers from the AMIG team at the Institut de Biologie Intégrative de la Cellule – I2BC (CNRS/CEA/UPSaclay, Gif-sur-Yvette) have shown that AlphaFold2 performs poorly if large disordered regions are used directly for prediction (40% success rate). A protein fragmentation strategy was found to be particularly well adapted to predicting the interfaces between folded domains and small protein motifs that fold on contact with the partner. Applied on a large scale using the Jean Zay HPC infrastructure on more than 900 complexes, this strategy achieved a success rate of almost 90%, a very encouraging result for the systematic screening of protein interaction networks. Nevertheless, the study calls for vigilance with regard to the risks of detecting false positives, which will be at the heart of future developments in artificial intelligence strategies such as AlphaFold2.
More information: https://www.nature.com/articles/s41467-023-44288-7
Contact: Jessica ANDREANI and Raphaël GUEROIS <jessica.andreani@i2bc.paris-saclay.fr> <raphael.guerois@i2bc.paris-saclay.fr>
The stringent response is strongly activated in the high antibiotic producer, Streptomyces coelicolor.
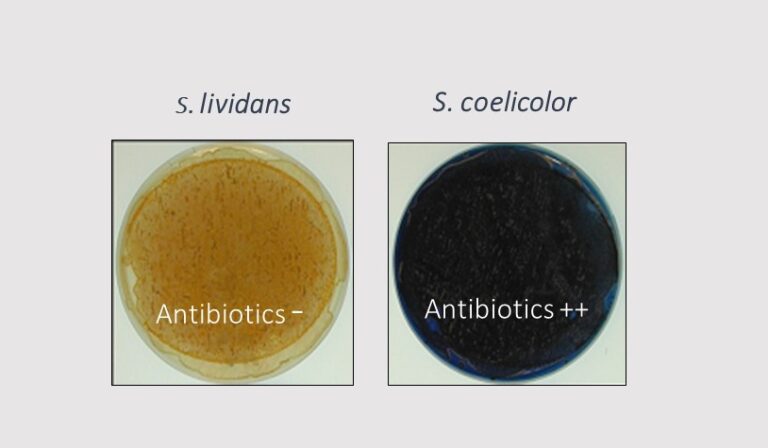
The stringent response controls positively antibiotic biosynthesis. Antibiotics are thus part of the stringent response.
In most bacteria, the stringent response (SR) was initially characterized as a response to nitrogen (N) limitation resulting into the depletion of aminoacylated tRNAs leading to the stalling of ribosomes on mRNA. The recruitment of the ppGpp synthetase, RelA, at the stalled ribosomes activates (p)ppGpp synthesis from GTP. ppGpp that is the mediator of the SR controls negatively, at the transcriptional and translational levels, the expression of most ribosomal proteins leading to the down regulation of the translational process and thus of growth.
The model strains Streptomyces coelicolor (SC) and Streptomyces lividans (SL), strong and weak producers of the same antibiotics, respectively, were grown in condition of phosphate (Pi) limitation or proficiency and the abundance of proteins of their translational apparatus was compared. This study revealed that the expression of RelA was induced in Pi limitation suggesting that, besides N limitation, Pi limitation also contributes to the triggering of the SR. Interestingly, most proteins of the translational apparatus had a similar or slightly higher abundance in SL than in SC, in Pi limitation whereas most of these proteins were far more abundant in SL than in SC, in Pi proficiency. This indicated an alleviation of the SR in Pi proficiency in SL, but not in SC. This suggested an alteration of Pi up-take and/or Pi-mediated regulation in SC whose molecular basis remain to be elucidated.
Interestingly, the production of specialized metabolites in SC (CDA, RED and ACT) is usually concomitant of phases of growth slow down and it is known that ppGpp controls positively the expression of their biosynthetic pathways. Their production could thus be considered as part of the SR. Indeed, these metabolites were proposed to regulate negatively, through different processes, the energetic metabolism and thus the generation of ATP, in SC, a process that might contribute to the slower growth rate of SC compared to SL.
More information: https://www.sciencedirect.com/science/article/pii/S0923250823001547?via%3Dihub
Contact: Marie-Joëlle VIROLLE <marie-joelle.virolle@i2bc.paris-saclay.fr>
Composition of Poxvirus Core Revealed

A collaboration between groups at I2BC, CSSB, MPI, and PEI, reveals the structure and the flexibility of A10 trimers that compose the palisade layer of the Vaccinia virus core encasing the viral genome.
Vaccinia virus is the model for the family of poxviruses, however, the structure of the viral particle used to propagate infection is poorly understood. The group of Emmanuelle Quemin at I2BC together with collaborators at CSSB in Hamburg, MPI for Biophysics in Frankfurt, and PEI in Langen, have revealed the composition and architecture of Vaccinia virus core that encases the viral genome. Their findings have been published in Nature Structural & Molecular Biology.
By combining cryo-electron tomography with subtomogram averaging and AlphaFold2, the authors were able to identify components of the core of Vaccinia virus. During entry, there is the fusion of the viral and cellular membranes that lead to the subsequent release of the viral core inside the host cell. As these are rare and fast events difficult to tackle by cellular cryo electron tomography, the researchers studied the core both in situ and in vitro, using virusal particles treated with detergents to access so-called “naked” cores containing the viral genome still. In parallel to the large dataset obtained in vitro, entering cores found inside cells were also analysed under native conditions at CSSB cryo-EM facility directed by Kay Grünewald in Hamburg, Germany,
The four groups involved focused more specifically on the outer layer of the core called the palisade that displayed a dense and organized surface of tubular protrusions that we refer to as stakes. Their study determined that these protrusions are trimers of the viral protein A10, previously known as one of the major core proteins. Here, the stakes appeared as more randomly organized than reported in other recent published work and have an inherent flexibility.
While some poxviruses can spread in human populations as recently exemplified with the Mpox virus multi-country outbreak, improving our understanding of Vaccinia virus and its core sub-structure is key to shed light on conserved mechanisms of poxvirus infection and pathogenicity.
More information: https://www.nature.com/articles/s41594-024-01218-5
Contact: Emmanuelle QUEMIN <emmanuelle.quemin@i2bc.paris-saclay.fr>
Schizosaccharomyces pombe as a fundamental model for research on mitochondrial gene expression: Progress, achievements and outlooks
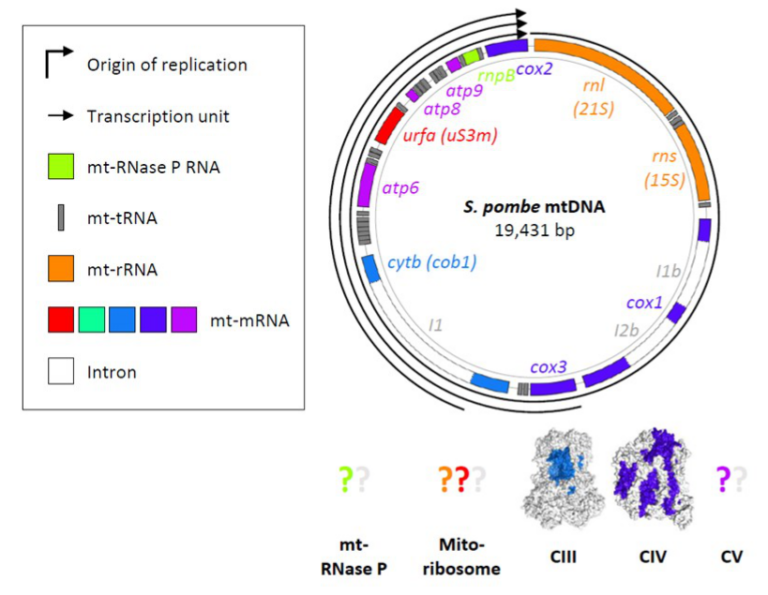
This is a comprehensive and critical review on mitochondrial gene expression in fission yeast, presenting up-to-date knowledge, and emphasising numerous contributions of the unicellular model to both fundamental and biomedical research.
Schizosaccharomyces pombe (fission yeast) is an attractive model for mitochondrial research. The organism resembles human cells in terms of mitochondrial inheritance, mitochondrial transport, sugar metabolism, mitogenome structure, and dependence of viability on the mitogenome (the petite-negative phenotype). Transcriptions of these genomes produce only a few polycistronic transcripts, which then undergo processing as per the tRNA punctuation model. In general, the machinery for mitochondrial gene expression is structurally and functionally conserved between fission yeast and humans. Furthermore, molecular research on S. pombe is supported by a considerable number of experimental techniques and database resources. Owing to these advantages, fission yeast has significantly contributed to biomedical and fundamental research. Here, we review the current state of knowledge regarding S. pombe mitochondrial gene expression, and emphasise the pertinence of fission yeast as both a model and tool, especially for studies on mitochondrial translation.
More information: https://doi.org/10.1002/iub.2801
Contact: Nathalie BONNEFOY <nathalie.bonnefoy@i2bc.paris-saclay.fr>
Antigen self-anchoring onto bacteriophage T5 capsid-like particles for vaccine design

Self-anchoring of large antigens onto Capsid-Like Particles derived from bacteriophage T5 paves the way for the development of a new vaccination platform that is highly immunogenic without the need for extrinsic adjuvant.
The constant need for immunization to prevent life-threatening diseases worldwide is urging the search for new vaccines. The promises of vaccines based on virus-like particles stimulate demand for universal non-infectious virus-like platforms that can be efficiently grafted with large antigens. In this study, we harnessed the modularity and extreme affinity of the decoration protein pb10 for the capsid of bacteriophage T5 to design a novel Ag delivery platform. DNA-free T5 capsid-like particles (T5-CLP) were decorated with chimeric proteins formed of pb10 fused to the model antigen ovalbumin (Ova). SPR experiments demonstrated that these proteins retained picomolar affinity for T5-CLP, while cryo-EM studies attested to the full occupancy of the 120 capsid binding sites. Mice immunisation with CLP-bound pb10-Ova chimeras elicited strong long-lasting anti-Ova humoral responses involving a large panel of isotypes, as well as CD8+ T cell responses, without any extrinsic adjuvant. Therefore, T5-CLP constitutes a unique DNA-free bacteriophage capsid able to display a regular array of large antigens through highly efficient chemical-free anchoring. Its ability to elicit robust immune responses paves the way for further development of this novel vaccination platform.
More information: https://www.nature.com/articles/s41541-023-00798-5
Contact: Pascale BOULANGER <pascale.boulanger-biard@i2bc.paris-saclay.fr>
Toxoplasma membrane inositol phospholipid binding protein TgREMIND is essential for secretory organelle function and host infection
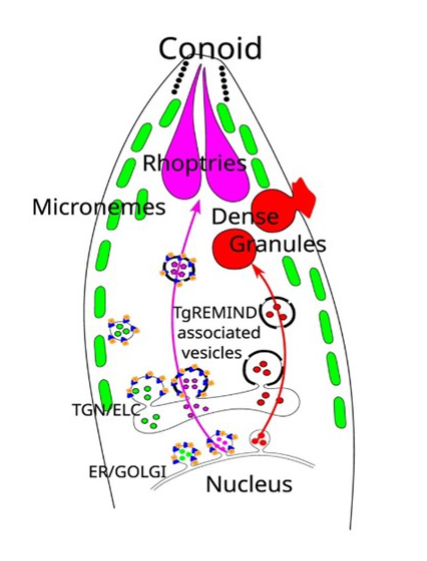
CApicomplexan parasites possess specialized secretory organelles called rhoptries, micronemes, and dense granules that play a vital role in host infection. In this study, we demonstrate that TgREMIND, a protein found in Toxoplasma gondii, is necessary for the biogenesis of rhoptries and dense granules. TgREMIND contains a Fes-CIP4 homology-Bin/Amphiphysin/Rvs (F-BAR) domain, which binds to membrane phospholipids, as well as a novel uncharacterized domain that we have named REMIND (regulator of membrane-interacting domain). Both the F-BAR domain and the REMIND are crucial for TgREMIND functions. When TgREMIND is depleted, there is a significant decrease in the abundance of dense granules and abnormal transparency of rhoptries, leading to a reduction in protein secretion from these organelles. The absence of TgREMIND inhibits host invasion and parasite dissemination, demonstrating that TgREMIND is essential for the proper function of critical secretory organelles required for successful infection by Toxoplasma.
More information: doi: 10.1016/j.celrep.2023.113601
Contact: Stanislas TOMAVO <stanislas.tomavo@i2bc.paris-saclay.fr>